
Abstract
Translation control of proinflammatory genes has a crucial role in regulating the inflammatory response and preventing chronic inflammation, including a transition to cancer. The proinflammatory tumor suppressor protein programmed cell death 4 (PDCD4) is important for maintaining the balance between inflammation and tumorigenesis. PDCD4 messenger RNA translation is inhibited by the oncogenic microRNA, miR-21. AU-rich element-binding protein HuR was found to interact with the PDCD4 3′-untranslated region (UTR) and prevent miR-21-mediated repression of PDCD4 translation. Cells stably expressing miR-21 showed higher proliferation and reduced apoptosis, which was reversed by HuR expression. Inflammatory stimulus caused nuclear-cytoplasmic relocalization of HuR, reversing the translation repression of PDCD4. Unprecedentedly, HuR was also found to bind to miR-21 directly, preventing its interaction with the PDCD4 3′-UTR, thereby preventing the translation repression of PDCD4. This suggests that HuR might act as a ‘miRNA sponge‘ to regulate miRNA-mediated translation regulation under conditions of stress-induced nuclear-cytoplasmic translocation of HuR, which would allow fine-tuned gene expression in complex regulatory environments.
Similar content being viewed by others

Introduction
Programmed cell death 4 (PDCD4) is a proinflammatory tumor suppressor gene that has an important role in maintaining the balance between inflammation and tumorigenesis. PDCD4 acts as a tumor suppressor by inhibiting neoplastic transformation, tumor progression and metastasis, and loss of PDCD4 function or expression has been found in many cancers.1 PDCD4 is induced by both apoptotic and inflammatory stimuli, promotes activation of the transcription factor nuclear factor kappa-B (NFκB) and suppresses the anti-inflammatory cytokine Interleukin 10.2 It acts as a translation repressor either by binding to the translation initiation factor eIF4a or by interacting with structured 5′-untranslated regions (UTRs) of specific messenger RNAs (mRNAs).3 PDCD4 expression is controlled by both transcriptional and post-transcriptional mechanisms. Post-transcriptional mechanisms that regulate mRNA stability and/or translation have important roles in regulating the expression of inflammatory genes by ensuring rapid and flexible control of inflammation initiation and resolution.4 Translational regulation is mediated by the signal-dependent binding of regulatory RNA-binding proteins (RBPs) or microRNAs (miRNAs) to specific sequence/structural elements in the 3′- and 5′-UTRs of mRNAs.5, 6
The translation of PDCD4 is inhibited by the miRNA miR-21 that binds to a single target site in the 3′-UTR of PDCD4 mRNA.7, 8, 9 miR-21 is an oncogenic miRNA (oncomiR) and increased levels of miR-21 have been found in a large number of malignancies and in several chronic inflammatory diseases.10 Increased miR-21 expression increases cell proliferation and inhibits apoptosis, whereas the inhibition of miR-21 causes tumor regression.11 miR-21 also acts as an important regulator of PDCD4 under inflammatory condition.12
Although a number of studies have shown the translation regulation of PDCD4 by miR-21, RBP-mediated regulation of PDCD4 translation has not been reported. Recently, three transcriptome-wide studies to identify the RNA-interactome of the RBP HuR indicated PDCD4 mRNA as one of the targets of HuR binding.13, 14, 15 HuR is an RBP belonging to the Hu/ELAV (embryonic lethal abnormal vision) family of RBPs, which interacts with AU-rich elements (AREs) mostly present in the 3′-UTR of target mRNAs to regulate stability and/or translation.16 Target mRNAs of HuR encode proteins involved in cell proliferation and differentiation, inflammation, angiogenesis, genotoxic and oxidative damage, hypoxia and nutrient deprivation.17 Interestingly, a number of recent reports have demonstrated functional interplay between HuR and specific miRNAs in regulating translation, where HuR binding to target mRNAs modulates miRNA-mediated repression of gene expression.18 However, the mechanism of the interplay between HuR and the miRNAs leading to translation regulation is not clearly understood.
In this study we have shown that HuR binds to the PDCD4 3′-UTR and inhibits miR-21-mediated translation repression in MCF7 breast carcinoma cells. A cell line stably expressing miR-21 showed higher rate of proliferation and reduced apoptosis, which was rescued by HuR expression. Treatment with an inflammatory agonist, bacterial lipopolysaccharide (LPS), caused the nuclear-cytoplasmic shuttling of HuR, resulting in the reversal of miR-21-mediated translation suppression of PDCD4. Remarkably, we found that HuR also binds to miR-21 directly, thereby preventing its interaction with the PDCD4 3′-UTR and suggest that HuR might act as a ‘miRNA sponge‘ to sequester miR-21 and prevent the translation repression of PDCD4.
Results
HuR binds to the PDCD4 mRNA 3′-UTR to reverse miR-21-mediated translation repression
miR-21 inhibits the translation of PDCD4 mRNA in MCF7 breast carcinoma cells by binding to a target site within the 3′-UTR9 (Supplementary Figure 1). We investigated whether HuR protein binds to the PDCD4 mRNA in MCF7 cells. Immunoprecipitation of HuR from MCF7 cell lysate, followed by reverse transcription-PCR, showed specific association of PDCD4 mRNA with HuR (Figure 1a). Further, immunoprecipitation of ultraviolet (UV)-crosslinked and RNase-A-digested RNA-protein complexes from binding reactions of MCF7 cell lysate, with radioactively labeled PDCD4 3′-UTR RNA, showed binding of HuR to the PDCD4 3′-UTR RNA (Figure 1b).

HuR binds to the PDCD4 mRNA 3′-UTR and reverses miR-21-mediated translation repression. (a) MCF7 cell lysates were immunoprecipitated with anti-HuR antibody or control IgG. RNA associated with the immunoprecipitate was subjected to RT–PCR using PDCD4 or GAPDH primers. (b) 32P-UTP-labeled PDCD4 3′-UTR RNA was incubated with MCF7 S10 cytoplasmic lysate, UV-crosslinked, digested with RNase-A. The RNase A digested reactions were immunoprecipitated with anti-HuR antibody or control IgG, and resolved on SDS-12% PAGE. The band corresponding to HuR is indicated by arrow. (c) Immunoblots of lysates of MCF7 cells transfected with two increasing concentrations of pSUP-miR-21 and co-transfected with two increasing concentrations of pCI-neo-myc-HuR together with the higher amount of pSUP-miR-21, probed with anti-PDCD4, anti-myc, anti-HuR and anti-GAPDH antibodies. The two bands in the anti-HuR immunoblot represent endogenous HuR and exogenously expressed myc-tagged HuR. Densitometric quantitation of PDCD4 protein bands, normalized to GAPDH, is given. (d) RT–PCR of total RNA isolated from MCF7 cells transfected with miR-21 and HuR-expressing plasmids with miR-21 and u6B snRNA primers. Densitometric quantitation of miR-21 PCR products, normalized to U6B snRNA, is given. (e) MCF7 cells transfected with Fluc-PDCD4 3′-UTR reporter gene construct and pCMV-Rluc were co-transfected with two increasing concentrations of pSUP-miR-21 and of three increasing concentrations of pCI-neo-myc-HuR in presence of the higher amount of pSUP-miR-21. Fluc values are normalized to Rluc values as transfection control. (f) MCF7 cells were either untransfected (-miR-21/-HuR) or transfected with pSUP-miR-21 together with (+miR-21/+HuR) or without pCI-neo-myc-HuR (+miR-21/-HuR) or with pCI-neo-myc-HuR alone (-miR-21/+HuR). Cell lysates were immunoprecipitated with anti-HuR antibody, anti-Ago2 antibody and control IgG. RNA associated with the immunoprecipitates was subjected to qRT–PCR using PDCD4 and GAPDH primers, and PDCD4 mRNA levels were normalized to GAPDH mRNA levels. Mean±s.d. from three experiments are represented in all graphs. * signifies a P-value⩽0.05 and ** signifies a P-value⩽0.01 (paired one-tailed t-test).
We then investigated the functional effect of HuR on miR-21-mediated translation repression of PDCD4. Overexpression of a short hairpin RNA corresponding to the miR-21 sequence inhibited PDCD4 expression in MCF7 cells in a dose-dependent manner (Figure 1c). However, overexpression of HuR, in presence of the highest concentration of miR-21, reversed the inhibition of PDCD4 expression. Exogenous expression of HuR did not reduce miR-21 level, suggesting that the effect of HuR was not mediated by degradation of the miRNA (Figure 1d). HuR expression also did not reduce the cellular level of argonaute 2 (Ago2), the major component of the translation-silencing RNA-induced silencing complex (RISC), suggesting that the effect of HuR was not mediated through the inactivation of RISC (Supplementary Figure 2a). HuR also did not inhibit miR-21 biogenesis or maturation as there was no significant change in either the level of precursor miR-21 or mature miR-21 on HuR overexpression (Supplementary Figure 2b). Also, the cellular level of PDCD4 mRNA remained unaffected by either miR-21 or HuR expression (Supplementary Figure 3). The effect of HuR was found to be mediated via the PDCD4 3′-UTR, as increased HuR expression reversed the miR-21-mediated repression of a reporter gene construct consisting of the PDCD4 3′-UTR inserted downstream of firefly luciferase gene (Fluc-PDCD4 3′-UTR) (Figure 1e). We also checked whether interaction with HuR could dissociate the PDCD4 mRNA from the RISC complex. PDCD4 mRNA was found to be associated with Ago2 by RNA-immunoprecipitation of lysates from cells expressing miR-21 (Figure 1f). Conversely, HuR overexpression resulted in significantly enhanced association of PDCD4 mRNA with HuR, suggesting that HuR expression could dissociate the PDCD4 mRNA from the RISC complex. Also, HuR overexpression resulted in significant reduction of miR-21 association with Ago2, suggesting that HuR led to dissociation of miR-21 from the RISC complex, and thereby from the PDCD4 mRNA (Supplementary Figure 4). Together these observations indicated that HuR could bind to the 3′-UTR of the PDCD4 mRNA and prevent miR-21-mediated translation silencing.
HuR reverses miR-21-mediated increase in cell proliferation and decrease in apoptosis
To investigate the cellular effects of the cross-talk between miR-21 and HuR, we generated a MCF7 cell line stably expressing miR-21, together with enhanced green fluorescent protein (EGFP) (MCF7-miR-21), which showed ~3.5-fold higher expression of miR-21 compared with a control cell line only expressing EGFP (MCF7-EGFP) (Supplementary Figure 5a). PDCD4 expression was strongly reduced in the MCF7-miR-21 cell line, whereas HuR level remained unaltered (Supplementary Figure 5b). Transfection of the Fluc-PDCD4 3′-UTR reporter gene construct in the cell line showed significant reduction in reporter gene expression, which was rescued by HuR overexpression (Supplementary Figure 5c). Polysome analysis showed that PDCD4 mRNA was mostly present in the non-translating lighter fractions of the MCF7-miR-21 cells compared with the MCF7-EGFP cells, in which it was mostly associated with the heavier polysomal fractions (Figure 2a). However, overexpression of HuR in the MCF7-miR-21 cells caused the shifting of PDCD4 mRNA to the polysomal fractions, showing that HuR could reverse the translation repression of PDCD4 caused by miR-21.

Derepression of PDCD4 translation by HuR counteracts miR-21-mediated increase in cell proliferation and decrease in apoptosis. (a) Ribosomal fractions from MCF7-EGFP, MCF7-miR-21 and MCF7-miR-21 cell line transfected with pCI-neo-myc-HuR were analysed by 10–50% sucrose density gradient fractionation. Ribosomal RNA content, measured at 254 nm, is plotted against fraction numbers. RNA isolated from selected fractions were analyzed by semiquantative RT–PCR using PDCD4 and GAPDH primers. (b) MCF7-EGFP and MCF7-miR-21 cell lines transfected with pCI-neo-myc-HuR or antagomiR against miR-21 or siRNA against HuR were allowed to grow post transfection and MTT assay was performed at various time points. OD595 readings are plotted. (c) MCF7-miR-21 cell line was transfected with pCI-neo-myc-HuR or antagomiR-21 and 48 h post transfection, DNA content of cells was analyzed by PI staining followed by flow cytometry. (d) MCF7-EGFP and MCF7-miR-21 cell lines transfected with pCI-neo-myc-HuR or an antagomiR against miR-21 or siRNA against HuR were serum-starved for 48 h post transfection. Caspase activation was measured using a caspase 3/7 assay using a luminescent substrate. RLU values are normalized to that of MCF7-EGFP cells, taken as 1. (e) MCF7 cells were either mock transfected, transfected with pSUP-miR-21 alone or together with pCI-neo-myc-HuR or HuR siRNA or with antagomiR-21 or HuR alone and then serum starved for 48 h to induce apoptosis. Cells were stained with AnexinV-FITC and PI to detect apoptosis. Mean±s.d. from three experiments done in duplicate are represented in all graphs. * signifies a P-value⩽0.05 and ** signifies a P-value⩽0.01 (t-test) in all graphs.
We also investigated whether the repression of PDCD4 expression could be rescued by HuR in a cell having high endogenous level of miR-21. The hepatocarcinoma cell line Huh7 had around threefold higher level of miR-21 compared with MCF-7, and although the PDCD4 mRNA level was around ninefold higher, the protein level was ~65% of that in MCF7 cells (Supplementary Figure 6a and b). Also, the HuR protein level in Huh7 cells was ~45% of that in MCF7 cells. This suggested that high level of miR-21, and low level of HuR, resulted in translation repression of PDCD4 in Huh7 cells even in the presence of high level of PDCD4 mRNA. Overexpression of HuR in the Huh7 cells significantly enhanced the expression of a Fluc-PDCD4 3′-UTR reporter gene construct, suggesting that HuR could reverse the translation repression of PDCD4 (Supplementary Figure 6c).
Comparison of proliferation rates of the MCF7-EGFP and MCF7-miR-21 cell lines over a 72 h period showed a significantly higher rate of proliferation of the miR-21 expressing cell line compared with the control (Figure 2b). Overexpression of HuR in the MCF7-miR-21 cells reduced the proliferation rate to that of control cells, an effect also shown by the transfection of an antagomiR to miR-21 (Figure 2b). Conversely, transfection of a HuR siRNA enhanced the proliferation rate (Figure 2b). The expression of HuR or antagomiR-21 in the MCF-7-miR-21 cell line showed a significant reduction in cells in S and G2/M phases, and an increase in cells in G0/G1 phase of the cell cycle (Figure 2c). Together, these results showed that miR-21 exerted an oncogenic effect by enhancing cell proliferation, which could be reversed by HuR expression or by blocking miR-21.
We also investigated the effect of miR-21 expression on apoptosis as translation silencing of PDCD4 by miR-21 reduces apoptosis. Caspase 3/7 activation in the MCF7-miR-21 cell line serum starved for 48 h to induce apoptosis was significantly lower compared with the MCF7-EGFP cell line (Figure 2d). However, the expression of HuR or antagomiR-21 significantly enhanced the caspase activation in the MCF7-miR-21 cell line, whereas the transfection of HuR siRNA reduced the caspase activity. Annexin V-FITC and propidium iodide staining of serum-starved cells showed high level of apoptosis, which was strongly reduced in cells transiently transfected with a miR-21 expressing construct and treated similarly (Figure 2e). The overexpression of HuR in the cells expressing miR-21 restored apoptosis, whereas transfection of HuR siRNA showed a strong reduction in apoptosis. HuR overexpression or transfection of antagomiR-21 also enhanced the early stages of apoptosis, marked by enhanced Annexin V staining. Together, these observations demonstrated that miR-21 suppressed apoptosis, whereas HuR could reverse the antiapoptotic effect of miR-21.
Treatment with LPS causes nuclear-cytoplasmic shuttling of HuR and prevents miR-21-mediated translation silencing of PDCD4
As PDCD4 is induced by inflammatory stimuli, we investigated the effect of LPS on regulation of HuR and its consequence on miR-21-mediated translation silencing of PDCD4. Treatment of MCF7 cells with LPS showed a time-dependent translocation of HuR from the nucleus to the cytoplasm, becoming mainly localized in the cytosol by ~4 h (Figure 3a). RNA-immunoprecipitation from LPS-treated cells with anti-HuR antibody showed fivefold higher association of HuR with PDCD4 mRNA 4 h after LPS treatment compared with untreated cells (Figure 3b). Similarly, LPS treatment of the MCF7-miR-21 cell line showed higher level of PDCD4 expression in comparison with untreated cells, which was abrogated on siRNA-mediated knockdown of HuR (Figure 3c). Polysome analysis of MCF7-miR-21 cells treated with LPS for 4 h also showed increased association of PDCD4 mRNA with the polysomal fractions in comparison with untreated MCF7-miR-21 cells shown earlier (Supplementary Figure 7a and Figure 2a). LPS treatment also enhanced PDCD4 and cytoplasmic HuR levels in Huh7 cells with low endogenous PDCD4 and HuR (Supplementary Figure 7b). Together, these observations suggested that treatment with an inflammatory agonist regulated HuR localization, and cytosolic localization of HuR resulted in binding to PDCD4 mRNA and reversal of miR-21-mediated translation silencing.

LPS treatment causes nuclear-cytoplasmic relocalization of HuR and prevents miR-21-mediated repression of PDCD4. (a) MCF7 cells were treated with LPS, and immunofluorescence of cells collected at various time points was observed using anti-HuR primary and AlexaFluor568-conjugated secondary antibodies. Nucleus was visualized using DAPI staining. Right panel shows merge of the DIC and fluorescent images. (b) Lysates of cells treated with LPS for 4 h were immunoprecipitated with anti-HuR antibody and control IgG. RNA associated with the immunoprecipitates was subjected to qRT–PCR using PDCD4 and GAPDH-specific primers, and PDCD4 mRNA levels were normalized to GAPDH mRNA levels. The data represent fold excess of normalized PDCD4 mRNA in HuR immunoprecipitate over IgG immunoprecipitate. Mean±s.d. from three experiments are represented. * signifies a P-value⩽0.05 and ** signifies a P-value⩽0.01 (t-test) (c). MCF-EGFP, MCF7-miR-21 and MCF7-miR-21 cell line transfected with HuR siRNA were treated with LPS for 4 h and cell lysates were immunoblotted using anti-PDCD4, anti-HuR and anti-β-actin antibodies. Densitometric quantitation of PDCD4 protein bands, normalized to β-actin, is given.
HuR binds to two sites in the PDCD4 3′-UTR in proximity to the miR-21 target site
We then tried to delineate the binding sites of HuR in the PDCD4 3′-UTR. Deletions of the PDCD4 3′-UTR, from the 5′ and 3′ ends, were generated in a stepwise manner (Figure 4a), and used in RNA-protein interaction assay to find the binding site(s) of HuR (Figure 4b). Deletion of 100 nucleotides from either the 3′ or 5′ ends (5′Δ1 and 3′Δ1), or of ~200 nucleotides from the 3′ end (3′Δ2) did not reduce HuR binding. However, removal of 200 nucleotides from the 5′ end (5′Δ2) significantly reduced, but did not abrogate, HuR binding. This suggested the presence of one HuR-binding site within the 5′ end 100–200 nucleotides of the PDCD4 3′ UTR. A deletion mutant lacking 100 nucleotides at the 5′ end, and 200 nucleotides at the 3′ end (5′Δ1–3′Δ2), did not show a reduction in HuR binding. However, when 200 nucleotides from the 5′ end and 300 nucleotides from the 3′ end were removed (5′Δ2–3′Δ3), there was complete abrogation of HuR binding. This suggested the presence of a second binding site between 350 and 450 nucleotides of the PDCD4 3′ UTR. Interestingly, the binding sites are located upstream and downstream of the miR-21 target site, with the sites present in close proximity, but non-overlapping with, the miR-21 target site. Competition UV-crosslinking using radiolabeled PDCD4 full-length 3′ UTR (FL) with three increasing concentrations of various unlabeled PDCD4 3′-UTR deletions also showed comparable binding to HuR for all the deletions, except the 5′Δ2–3′Δ3 fragment, which failed to show any competition for HuR binding (Supplementary Figure 8). This indicated the absence of HuR-binding sites within the 200–350 nt region of the PDCD4 3′-UTR.

HuR acts in trans to prevent miR-21 mediated translation repression of PDCD4. (a) Schematic diagram of full-length (FL) PDCD4 3′-UTR and various deletions from the 5′- and 3′-ends. The miR-21 target site (nt 228–249) as well as the HuR-binding sites, as determined from the RNA-protein interaction studies (below), are indicated. (b) 32P-UTP labeled full-length and various deletion mutants of PDCD4 3′-UTR RNA were incubated with purified HuR protein, UV-crosslinked, digested with RNase A and resolved on 10% SDS–PAGE. (c) Reporter gene constructs containing full-length and various deletion mutants of PDCD4 3′-UTR were transfected into MCF cells together with pSUP-miR-21 or pCI-Neo-myc-HuR or both. (d) Reporter gene constructs containing full-length or various mutants (HuR binding and miR-21 binding) of the PDCD4 3′-UTR were co-transfected with pSUP-miR-21 vector and treated with LPS for 4 h. Mean±s.d. of Fluc/Rluc values, normalized to control, from three independent experiments done in duplicate are represented in all graphs. * signifies a P-value⩽0.05 and ** signifies a P-value⩽0.01 (t-test) in all graphs.
HuR acts in trans to prevent miR-21-mediated translation silencing of PDCD4
To decipher the functional role of the HuR-binding sites in the rescue of miR-21-mediated translation repression induced by HuR, we made reporter gene constructs containing deletion fragments of the PDCD4 3′-UTR lacking either or both HuR-binding sites. The presence of either of the upstream or downstream HuR-binding sites (5′Δ2–3′Δ2 and 5′Δ1–3′Δ3) was sufficient to rescue the miR-21-mediated translation repression by HuR (Figure 4c). Also, the expression of HuR alone showed moderate enhancement of luciferase expression, supporting the role of HuR in upregulating translation. However, remarkably, even when both the HuR-binding sites were removed (5′Δ2–3′Δ3), HuR expression caused significant reversal of miR-21-mediated translation inhibition. This suggested that HuR could also act in trans, without directly interacting with the 3′-UTR, to prevent miR-21-mediated translation silencing of PDCD4 mRNA. Treatment with LPS also reversed the miR-21-mediated translation repression of the reporter gene construct containing the PDCD4 3′-UTR 5′Δ2–3′Δ3 sequence, whereas there was no translation repression from a reporter gene construct containing a PDCD4 3′-UTR where the seed sequence of the miR-21-binding site has been mutated (Figure 4d). LPS treatment or miR-21 overexpression did not affect the RNA levels of the luciferase reporter gene constructs (Supplementary Figure 9).
HuR interacts with miR-21 independently of PDCD4 mRNA
The ability of HuR to rescue the miR-21-mediated translation repression of PDCD4 without binding to the PDCD4 3′-UTR suggested that HuR might directly interact with miR-21 and prevent its binding to the PDCD4 3′-UTR. Interestingly, the miR-21 sequence showed the presence of a non-canonical ARE, UAUU, within the seed sequence, which could be a potential binding site for HuR (Figure 5a). RNA electrophoretic mobility shift assay using fluorescently labeled miR-21 RNA, miR-125b RNA, which lacks any ARE, a non-specific RNA of equal length and a miR-21 RNA where the ARE sequence has been mutated to CGCG, showed that only wild-type miR-21 RNA could bind with HuR, whereas no binding was observed in case of miR-125b or the non-specific RNA or ARE-mutant miR-21 RNA (Figure 5b). This suggested that the binding of HuR to the miR-21 RNA is specific and mediated through the ARE. A non-specific protein (bovine serum albumin) also failed to show any binding to the miR-21 RNA.

HuR binds directly with miR-21. (a) Sequence of miR-21 (guide strand) and target site in PDCD4 3′-UTR with the ARE within the seed sequence of miR-21 indicated. (b) Cy5-labeled fluorescent RNA corresponding to miR-21, miR-125b, ARE-mutated miR-21, a non-specific RNA of equivalent length were incubated with purified HuR protein and the RNA-protein complex resolved by native PAGE. miR-21 was also incubated with purified BSA followed by native PAGE. (c) Isothermal Titration Calorimetry (ITC) was performed using in vitro transcribed miR-21 RNA as analyte and purified HuR protein as titrant. Heat change on injection of titrant was measured (upper panel) and heat change per mole of titrant was plotted against the molar ratio of titrant to analyte. (d) Filter binding assay using 32P-ATP end-labeled RNA corresponding to miR-21, ARE-mutant miR-21, double-stranded miR-21, miR-125b and a non-specific RNA of equivalent length and purified HuR protein. Only single-stranded miR-21 (blue line) showed significant binding. miR-125b and non-specific RNA did not show any binding at all. The miR-21 binding curve was fitted using a polynomial function and the Kd was computed from the curve. Mean±s.d. from three experiments are represented.
RNA-protein interaction studies by isothermal titration calorimetry and filter-binding assay showed interaction between miR-21 and HuR (Figures 5c and d), whereas there was no significant interaction with miR-125b or the non-specific RNA (Supplementary Fig 10a). The ARE-mutant miR-21 RNA and a double-stranded miR-21 RNA failed to show binding with HuR in the filter-binding assay. This suggested that HuR could bind specifically to the single-stranded miR-21 RNA via the non-canonical ARE. The affinity of HuR to miR-21 was found to be in the micromolar range, with a Kd of ~1 μM, suggesting transient interaction between HuR and miR-21 that could take place only in presence of high concentration of HuR in cells.
To check the interaction of HuR with miR-21 in cells, HuR was overexpressed in the MCF7-EGFP and the MCF7-mir-21 cell lines, followed by immunoprecipitation using anti-HuR antibody and quantitative reverse transcription-PCR for miR-21 (Figure 6a). miR-21 was found to be significantly enriched in the HuR immunoprecipitate in both cell lines with an approximately fivefold enrichment in the miR-21 expressing cell line. Conversely, PDCD4 mRNA was found to be significantly associated with HuR in the MCF7-EGFP cell line and showed insignificant association with HuR in the MCF7-miR-21 cell line (Figure 6b). LPS treatment for 4 h also significantly enhanced association of miR-21 with HuR (Figure 6c). HuR was also found to specifically interact with the mature miR-21, but not with the precursor miR-21 in cells (Supplementary Figure 10b)

HuR binds to miR-21 in cells independently of PDCD4 mRNA. (a) Lysates from miR-21 and EGFP-expressing cell lines in which HuR protein was overexpressed were immunoprecipitated with anti-HuR and control IgG antibodies. qRT–PCR was done with immunoprecipitated RNA using miR-21 and U6B snRNA-specific primers. (b) Lysates from miR-21 and EGFP-expressing cell lines were immunoprecipitated with anti-HuR and control IgG antibodies followed by qRT–PCR using PDCD4 and GAPDH-specific primers. (c) MCF7 cells were treated with LPS for 4 h, followed by immunoprecipitation with anti-HuR and control IgG antibodies and qRT–PCR using miR-21 and U6B snRNA-specific primers. (d) Lysates from MCF7 cells in which PDCD4 3′-UTR RNA has been overexpressed were immunoprecipitated with anti-HuR and control IgG antibodies followed by qRT–PCR using miR-21 and U6B-specific primers. The data show fold difference in miR-21 levels (normalized to U6B snRNA levels) in the HuR immunoprecipitates compared with IgG immunoprecipitates in panels a, c and d and PDCD4 mRNA (normalized to GAPDH levels) in panel b. (e) Lysates from MCF7 cells were immunoprecipitated with anti-HuR and anti-Ago2 antibodies and the immunoprecipitates resolved on SDS–PAGE and blotted with anti-HuR and anti-Ago2 antibodies (upper panel). Quantative RT–PCR was performed with RNA isolated from the immunoprecipitates using miR-21 and U6B snRNA-specific primers. The data show fold difference in miR-21 levels (normalized to U6B snRNA levels) in the HuR and Ago2 immunoprecipitates compared with IgG immunoprecipitates (lower panel). (f) MCF7 cells were treated with LPS for 0–4 h, followed by immunoprecipitation with anti-HuR and control IgG antibodies and qRT–PCR using miR-21 and U6B snRNA-specific primers. Fold difference in miR-21 level (normalized to U6B level) in HuR immunoprecipitate compared with IgG immunoprecipitate is shown. The cell lysates were immunoblotted with anti-PDCD4 and GAPDH antibodies (lower panel). Densitometric quantitation of PDCD4 protein bands, normalized to GAPDH, is given. Mean±s.d. from three experiments are represented in all graphs. * signifies a P-value⩽0.05 and ** signifies a P-value⩽0.01 (t-test) in all graphs.
We also investigated the role of the PDCD4 3′-UTR in the interaction between HuR and miR-21. The overexpression of PDCD4 3′-UTR RNA in cells showed increased association of HuR with miR-21 (Figure 6d), suggesting that increased level of PDCD4 3′-UTR RNA enhanced the interaction between miR-21 and HuR. To determine whether the interaction between miR-21 and HuR reflected a three-way interaction between miR-21, PDCD4 mRNA and miR-21-RISC, we immunoprecipitated HuR in the absence of RNase treatment and checked for association with Ago2 and vice-versa (Figure 6e, upper panel). HuR and Ago2 did not show any interaction, suggesting that HuR and the miR-21-RISC complex were not binding simultaneously to the PDCD4 mRNA. However, both HuR and Ago2 showed interaction with miR-21 (Figure 6e, lower panel). Together these observations showed that HuR could bind to miR-21, independent of the PDCD4 3′-UTR RNA.
To further probe the mechanism of interactions between miR-21, PDCD4 3′-UTR and HuR, we designed miR-21 seed sequence mutant of the PDCD4 3′-UTR, which fail to bind to miR-21, and miR-21 seed sequence mutants, which fail to bind to HuR but will bind to the mutant PDCD4 3′-UTR (Supplementary Figure 11a). Reporter gene constructs containing full-length PDCD4 3′-UTR (FL), full-length PDCD4 3′-UTR in which the miR-21 target site seed sequence has been mutated (miR-21ss-mut) and PDCD4 3′-UTR, which does not bind to HuR (5′Δ2–3′Δ3) were co-transfected into MCF7 cells with miR-21-expressing constructs, which either expressed wild-type miR-21 (WT) or miR-21 in which the seed sequence, including the ARE, has been mutated (ss-mut) so that it compensates for the seed sequence mutation in the PDCD4 3′-UTR but will fail to bind to HuR. Luciferase reporter assay showed that the seed sequence mutated miR-21 could bind to the compensatory seed sequence mutated PDCD4 3′-UTR and repress translation (Supplementary Figure 11b). miR-21(ss-mut) expressed well in cells upon transfection and was detectable using specific primers (Supplementary Figure 11c). HuR was immunoprecipitated from the transfected cells, followed by quantitative reverse transcription-PCR using specific primers for WT miR-21 and the mutant miR-21. HuR was found to bind to WT miR-21, but failed to bind to the mutant miR-21. Interestingly, even when the seed sequence mutated PDCD4, 3′-UTR was co-transfected with the compensatory seed sequence mutated miR-21, HuR failed to bind to the mutant miR-21, although the mutant miR-21 was able to bind to the PDCD4 3′-UTR and repress translation. Together these observations showed that HuR could bind to miR-21, independent of the PDCD4 3′-UTR RNA, and thereby prevent miR-21-mediated translation repression of PDCD4.
LPS-induced cytoplasmic translocation of HuR increases miR-21 binding and enhances PDCD4 expression
LPS treatment causes nuclear-cytoplasmic translocation of HuR, accompanied with increased binding to PDCD4 mRNA and rescue of PDCD4 translation (Figure 3). We now investigated whether the nuclear-cytoplasmic shuttling of HuR (Supplementary Figure 12) on LPS treatment also enhances its interaction with miR-21. RNA-immuoprecipitation using anti-HuR antibody from LPS-treated cell lysates showed a time-dependent increase in miR-21 interaction, with a significant increase at 4 h post LPS treatment (Figure 6f). This was accompanied by the highest level of PDCD4 protein expression. This suggested that the gradual cytoplasmic localization of HuR on LPS treatment resulted in a ‘sponging‘ of miR-21 by HuR, leading to the reversal of miR-21-mediated PDCD4 translation repression.
Discussion
In this study we have described a new mode of regulation of PDCD4 mRNA translation mediated by an antagonistic interplay between the microRNA miR-21 and the RBP HuR. Multiple examples of interplay between RBPs and miRNAs, both competitive and cooperative, in the regulation of gene expression are now known. In the first reported case, HuR translocation from nucleus to cytoplasm under amino-acid deprivation prevented the miR-122-mediated repression of cationic amino-acid transporter CAT-1 mRNA.19 Similar competitive cross-talk has been reported between HuR and miRNAs such as miR-548c, miR-494, miR-16, miR-331-3p and miR-1192 in response to different signals.20, 21, 22, 23, 24 Interplay between RBPs and miRNAs appear to be an important mode of fine-tuning gene expression in various processes related to cancer. For instance, a complex consisting of hnRNP L binds to CA-rich sequences in the hypoxia stability region of vascular endothelial growth factor-A mRNA in response to hypoxia and prevent the binding of both miR-297/299 and the translation silencing interferon gamma activated inhibitor of translation complex, thereby allowing the expression of vascular endothelial growth factor-A and angiogenesis.25, 26, 27 Various other RBPs such as tristetraprolin, hnRNP E2 and IGF2BP1 have similar roles in the context of multiple oncogenes and tumor suppressor genes.28
Although multiple instances of cross-talk between miRNAs and RBPs in the regulation of translation/mRNA stability is now known, the mechanism of such interplay is still not well understood. Antagonistic interplay between RBPs and miRNAs might involve direct steric hindrance, where the binding sites are overlapping such as in the case of miR-16 and HuR binding to the COX-2 mRNA.23 However, the high-throughput studies to analyze the mRNA interactome of HuR has predicted most miRNA-binding sites to be non-overlapping, but in the vicinity of HuR-binding sites.14, 15 Similarly, in the PDCD4 3′-UTR, the HuR-binding sites are non-overlapping with the miR-21-binding site, suggesting long-range interaction between HuR and miR-21 binding. Such long-range interaction might be mediated by RNA conformational alterations where the binding of the RBP causes a conformational change in the RNA structure to make the miRNA target site more or less accessible to the mi-RISC complex, such as in the case of pumilio-mediated regulation of miR-221/222 binding to p27 mRNA.29 In the case of PDCD4 mRNA, binding of HuR inhibits miR-21 binding to the target site, thereby causing translation derepression. Transcriptome-wide analyses of HuR target mRNAs have predicted ~1700 mRNAs, which have both HuR and miRNA-binding sites in their 3′-UTR.13, 14 We found that out of these 1700 mRNAs, 109 were also present among the 307 predicted targets of miR-21 with conserved seed sequences (Targetscan, www.targetscan.org) (Figure 7a). Therefore, at least 109 mRNAs are potential targets of co-regulation by HuR and miR-21, suggesting a large post-transcriptional co-regulon responsive to both miR-21 and HuR.

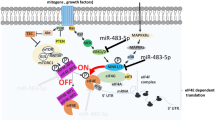
Integration of miR-21 and HuR-mediated regulation and model of the ‘miRNA sponge‘. (a) Intersection of mRNAs having HuR and miRNA-binding sites with predicted miR-21 target mRNAs. The intersection of 109 mRNAs represent the co-regulon of HuR and miR-21. (b) Pie-chart showing distribution of various AREs and HuR-binding sites in miRNA sequences from miRbase. Total number of miRNAs with ARE sequences or HuR-binding sites is 672 and total number of AREs and HuR-binding sites is 1245. (c) Frequency distribution showing number of AREs per miRNA sequence. (d) Proposed model showing dynamic equilibrium between miR-21 and HuR binding to PDCD4 mRNA and ‘miRNA sponge‘ function of HuR. High level of miR-21 allows miRNA-RISC complex to be loaded on PDCD4 mRNA 3′-UTR and prevents HuR binding to the 3′-UTR, causing translation repression. High level of HuR in the cytoplasm sequesters miR-21, and causes conformational change in the PDCD4 3′-UTR, causing more of miR-21 to dissociate from PDCD4 mRNA 3′-UTR and RISC complex, leading to translation derepression.
Moreover, in the case of PDCD4, a remarkable observation was that HuR could inhibit miR-21-mediated translation repression by directly binding and sequestering miR-21. In some recent studies, HuR has been reported to interact with miRNAs such as miR-16, miR-1192 and mir-29 to regulate the expression of COX-2, HMGB1 and A20 genes, respectively.23, 24, 30 In the case of miR-21, our observations suggest that HuR binds single stranded, cytoplasmic miR-21, acting as a ‘miRNA sponge’, and prevents translation repression of PDCD4. Non-coding RNAs, transcribed from pseudogenes, which have multiple binding sites for specific miRNAs, have been reported to act as ‘miRNA sponges’ by sequestering miRNAs and causing derepression of their target genes.31 Conversely, miR-328 has been reported to act as an RNA decoy by binding and sequestering an RBP, hnRNP E2, in leukemic blasts.32 Our study leads us to hypothesize HuR as the first protein ‘miRNA sponge‘, which sequesters miR-21 in the cytoplasm and causes translational derepression of its target gene PDCD4. in vitro interaction studies appear to indicate a low affinity between HuR and miR-21 (Kd~1 μM), suggesting that such an interaction will be biologically relevant only in presence of high concentration of HuR. Such high concentrations of HuR might be transiently attained by nuclear-cytoplasmic relocalization of HuR, known to be caused by multiple stimuli such as LPS, heat shock and UV irradiation.33, 34 Although HuR is a highly abundant protein, being among the top third of proteins present in cells in terms of quantity,15 the hypothesis can be definitively proved only on accurate in vivo measurement of HuR and miR-21 in the cytoplasm on HuR relocalization. Generalized miRNA binding by RBPs such as HuR has not been envisaged, as the latter are thought to be mainly involved in mRNA binding. However, bioinformatic analysis of miRNA sequences showed the presence of ~1250 AREs and other HuR-binding sites reported in literature24, 30 and in the RBP database (http://rbpdb.ccbr.utoronto.ca/) in 672 miRNAs, with ~350 miRNAs having a single ARE sequence (Figures 7b and c). This suggests that multiple miRNAs might be targets of HuR, and possibly other RBP binding, which might constitute a more generalized mode of gene regulation.
Based on our observations we propose a model of regulation of PDCD4 mRNA translation in which HuR, on cytoplasmic localization, can bind to both the PDCD4 3′-UTR RNA and free, cytoplasmic miR-21 (Figure 7d). Free miR-21 is in dynamic equilibrium with the miR-21 loaded on to the PDCD4 3′-UTR in complex with RISC. Binding of HuR to the PDCD4 3′-UTR causes a conformational change, which reduces the affinity of miR-21 binding to its target site and causes its dissociation from the mRNA and the RISC complex. As the single stranded, cytoplasmic miR-21 is ‘sponged‘ up by HuR, the equilibrium between mRNA-bound and free miR-21 shifts more toward free miR-21 and causes further dissociation of miR-21 from the PDCD4 mRNA. Therefore, HuR binding to miR-21 and to the PDCD4 3′-UTR acts in concert, causing a dynamic equilibrium shift in miRNA-RISC-mRNA interaction, leading to translation derepression of PDCD4 mRNA. Discovery of the miRNA interactome of HuR, together with elucidation of the conformational changes in the mRNA 3′-UTRs on HuR and miRNA interactions, will lead to deeper understanding of this new mode of translation regulation and its role in tumorigenesis.
Materials and methods
Plasmid constructs
The 642 nt long human PDCD4 3’-UTR was amplified by reverse transcription-PCR from human leukocyte RNA and cloned into pCDNA3.1 (Life Technologies, Carlsbad, CA, USA) and pCDNA3-FLuc vector downstream of firefly luciferase (Fluc) gene. Different deletion constructs were generated by PCR from the pCDNA3.1-PDCD4 3’-UTR construct. Mutations in the PDCD4 3’-UTR miR-21-binding site seed sequence was incorporated by megaprimer-based method. Double-stranded DNA oligo encoding wild-type or mutated miR-21 sequence was cloned into pSUPER vector (Oligoengine, Seattle, WA, USA), which was transcribed to produce short hairpin RNA corresponding to miR-21. HuR was expressed from the mammalian expression vector pCI-neo-HuR (gift from S Bhattacharyya, CSIR-IICB) and from the bacterial expression vector pET28b-HuR (gift from S Das, IISc).
Cell culture, treatment and transfection
MCF7 human breast carcinoma cells and Huh7 hepatocarcinoma cells were maintained in Dulbecco's Modified Eagle Medium (Life Technologies) and RPMI1640 media, respectively, supplemented with 10% fetal bovine serum. Cells were treated with 100 ng/ml LPS from Escherichia coli (Sigma-Aldrich, St Louis, MO, USA). Cells were transfected with plasmid vectors, siRNA or antagomiRs using Lipofectamine 2000 (Life Technologies) in serum-free OptiMEM medium. DNA amount for transfection was equalized by pGEMT plasmid (Promega, Madison, WI, USA).
Generation of stable cell lines
The Lenti-PAC HIV Expression system (Genecopoeia Inc., Rockville, MD, USA) was used to generate MCF7 cell line stably expressing miR-21 short hairpin RNA according to manufacturer’s protocol. In brief, HEK293T cells were transfected with lentiviral vectors, together with viral packaging vectors. Pseudoviral particles obtained from the cell supernatants were used to transduce MCF7 cells. Clonal populations of transduced cells expressing EGFP were selected in presence of 1 μg/ml puromycin to generate stable cell lines.
RNA-protein UV-crosslinking assay
In vitro transcribed α-32P-UTP-labeled RNAs of equal specific activity were UV-crosslinked with purified protein or cell lysate and RNase-A digested as described earlier.35 The RNA-protein complexes were resolved on SDS–PAGE followed by phosphorimaging (Typhoon Trio, GE Healthcare, Pittsburgh, PA, USA). For competition UV-crosslinking assays, protein was incubated with increasing concentrations of unlabeled competitor RNAs before incubation with radiolabeled RNA, followed by UV-crosslinking, RNase digestion and gel electrophoresis.
RNA electrophoretic mobility shift assay
Fluorescent Cy-5-UTP-labeled RNA was used for RNA electrophoretic mobility shift assay. Labeled RNA was incubated with purified HuR protein in RNA-binding buffer on ice. The reaction mixture was resolved on 8% native PAGE and fluorescent bands were visualized using Typhoon Trio multimode imager (GE Healthcare).
Immunoblotting
Equal amounts of cell lysates were resolved by 12% SDS–PAGE, electro-transferred to polyvinylidene difluoride membrane followed by immunoblotting using antibodies against PDCD4 (Cell Signaling Technology, Danvers, MA, USA), HuR (Santacruz Biotechnology, Dallas, TX, USA), GAPDH (Santacruz Biotechnology), myc (Cell Signaling Technology) and horseradish peroxidase-conjugated β-Actin (Genscript, Piscataway, NJ, USA) or anti-Mouse (Cell Signaling Technology) or anti-Rabbit (Cell Signaling Technology) secondary antibodies. The bands were detected using Femtolucent chemiluminescence detection kit (G-Biosciences, St Louis, MO, USA).
RNA immunoprecipitation
Pre-cleared cell lysate was incubated with antibody (1:100 dilution) in radioimmunoprecipitation assay buffer or NT2 buffer for 4 h at 4 ºC. Pre-swelled Protein A sepharose beads (Sigma-Aldrich) were added to the antibody-protein complex and kept for overnight at 4 ºC. Unbound proteins were removed by washing with radioimmunoprecipitation assay buffer or NT2 buffer. RNA was isolated from the beads using TRIzol reagent (Life Technologies) and complementary DNA was prepared using MMLV Reverse Transcriptase (Fermentas, Thermo Fisher Scientific, Waltham, MA, USA).
miRNA and mRNA quantification by real-time PCR
Total RNA was isolated from cells using TRIzol Reagent and used for reverse transcription and quantitative PCR. Precursor and mature miRNA quantification was done using miScript PCR assay kit (Qiagen, Hilden, Germany) following manufacturer’s protocol. mRNA quantification was done using reverse transcription with oligo dT primer and qPCR using gene-specific primers. RNA quantity normalization between samples for ΔCt calculation was done using U6B snRNA-specific primers for miRNA quantification and GAPDH primers for mRNA quantification. qPCR was performed using SYBR Green reagent (Applied Biosystems, Life Technologies), in Step One plus real-time PCR system (Life Technologies).
Cell-proliferation and apoptosis assay
Proliferation of cell lines transfected with plasmid constructs/siRNA/antagomiR was measured by MTT assay (Sigma-Aldrich). For cell cycle analysis, 48 h after transfection, cells were harvested, fixed and stained with propidium iodide. DNA content was analysed using flow cytometry (BD FACScaliber). For apoptosis assay, stable cell lines were transfected and then serum starved (Dulbecco's Modified Eagle Medium with 2% fetal bovine serum) for 48 h. Apoptosis was detected by Annexin V-FITC/PI Apoptosis Detection Kit (Sigma-Aldrich) or by caspase assay using CaspaseGlo caspase 3/7 assay kit (Promega) following manufacturer’s protocol.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde and treated with 1:200 diluted mouse monoclonal anti-HuR antibody (Santa Cruz Biotechnology) followed by 1:400 diluted Alexafluor 568-conjugated rabbit anti mouse antibody (Cell Signaling Technology) and DAPI (0.5 ng/ml) as nuclear stain. Images were taken using Zeiss LSM 710 scanning confocal microscope.
Polysome analysis
Ribosomal fractions were obtained as described earlier.25 in brief, cells were homogenized in polysome lysis buffer containing cycloheximide (0.1 mg/ml) and cytosolic extract was obtained by centrifugation at 10 000 g for 20 min. The extract was overlayed on a 10–50% (w/v) sucrose gradient and centrifuged at 100 000 g for 4 h. Fractions were collected using a programmable gradient fractionator (Biocomp, Fredericton, NB, Canada) and absorbance at 254 nm was measured. RNA was isolated from the fractions by phenol-CHCl3 extraction and ethanol precipitation.
RNA-protein filter-binding assay
γ32P-ATP-labeled RNAs of equal specific activities were incubated with purified HuR protein in RNA-binding buffer on ice. Reaction mixtures were spotted on D80 nitrocellulose membrane, air dried, washed twice with RNA-binding buffer and once with cold ethanol in a vacuum trap. The radioactivity retained on the membranes was measured by scintillation counter.
Isothermal titration calorimetry
Isothermal titration calorimetry to study RNA-protein interaction was performed in a Microcal iTC 200 system (GE Healthcare). in vitro transcribed RNA was taken in RNA-binding buffer as analyte and 20-fold excess of purified HuR protein was taken as titrant. Stepwise injections of equal volumes of protein were done into the cell and heat changes were measured. The heat change data were plotted against molar ratio of titrant to analyte and curve fitting was done using a one-site model with reiterative χ2-test.
References
Lankat-Buttgereit B, Göke R . The tumour suppressor Pdcd4: recent advances in the elucidation of function and regulation. Biol Cell 2009; 101: 309–317.
Hilliard A, Hilliard B, Zheng S-J, Sun H, Miwa T, Song W et al. Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J Immunol 2006; 177: 8095–8102.
Biyanee A, Ohnheiser J, Singh P, Klempnauer K-H . A novel mechanism for the control of translation of specific mRNAs by tumor suppressor protein Pdcd4: inhibition of translation elongation. Oncogene 2014; 34: 1384–1392.
Anderson P . Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol 2010; 10: 24–35.
Gebauer F, Hentze MW . Molecular mechanisms of translational control. Nat Rev Mol Cell Biol 2004; 5: 827–835.
Filipowicz W, Bhattacharyya SN, Sonenberg N . Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 2008; 9: 102–114.
Lu Z, Liu M, Stribinskis V, Klinge CM, Ramos KS, Colburn NH et al. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008; 27: 4373–4379.
Asangani IA, Rasheed SAK, Nikolova DA, Leupold JH, Colburn NH, Post S et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008; 27: 2128–2136.
Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH . Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem 2008; 283: 1026–1033.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D et al. MicroRNA expression profiles classify human cancers. Nature 2005; 435: 834–838.
Si M-L, Zhu S, Wu H, Lu Z, Wu F, Mo Y-Y . miR-21-mediated tumor growth. Oncogene 2007; 26: 2799–2803.
Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol 2010; 11: 141–147.
Uren PJ, Burns SC, Ruan J, Singh KK, Smith AD, Penalva LOF . Genomic analyses of the RNA-binding protein Hu antigen R (HuR) identify a complex network of target genes and novel characteristics of its binding sites. J Biol Chem 2011; 286: 37063–37066.
Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M et al. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples Pre-mRNA processing and mRNA stability. Mol Cell 2011; 43: 327–339.
Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M et al. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell 2011; 43: 340–352.
Hinman MN, Lou H . Diverse molecular functions of Hu proteins. Cell Mol Life Sci 2008; 65: 3168–3181.
Abdelmohsen K, Kuwano Y, Kim HH, Gorospe M . Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol Chem 2008; 389: 243–255.
Srikantan S, Tominaga K, Gorospe M . Functional interplay between RNA-binding protein HuR and microRNAs. Curr Protein Pept Sci 2012; 13: 372–379.
Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W . Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 2006; 125: 1111–1124.
Srikantan S, Abdelmohsen K, Lee EK, Tominaga K, Subaran SS, Kuwano Y et al. Translational control of TOP2A influences doxorubicin efficacy. Mol Cell Biol 2011; 31: 3790–3801.
Tominaga K, Srikantan S, Lee EK, Subaran SS, Martindale JL, Abdelmohsen K et al. Competitive regulation of nucleolin expression by HuR and miR-494. Mol Cell Biol 2011; 31: 4219–4231.
Epis MR, Barker A, Giles KM, Beveridge DJ, Leedman PJ . The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J Biol Chem 2011; 286: 41442–41454.
Young LE, Moore AE, Sokol L, Meisner-Kober N, Dixon DA . The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol Cancer Res 2012; 10: 167–180.
Dormoy-Raclet V, Cammas A, Celona B, Lian XJ, van der Giessen K, Zivojnovic M et al. HuR and miR-1192 regulate myogenesis by modulating the translation of HMGB1 mRNA. Nat Commun 2013; 4: 2388.
Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL . A stress-responsive RNA switch regulates VEGFA expression. Nature 2009; 457: 915–919.
Jafarifar F, Yao P, Eswarappa SM, Fox PL . Repression of VEGFA by CA-rich element-binding microRNAs is modulated by hnRNP L. EMBO J 2011; 30: 1324–1334.
Yao P, Potdar AA, Ray PS, Eswarappa SM, Flagg AC, Willard B et al. The HILDA complex coordinates a conditional switch in the 3’-untranslated region of the VEGFA mRNA. PLoS Biol 2013; 11: e1001635.
Van Kouwenhove M, Kedde M, Agami R . MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer 2011; 11: 644–656.
Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JAF, Elkon R, Agami R . A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol 2010; 12: 1014–1020.
Balkhi MY, Balkhi MY, Iwenofu OH, Bakkar N, Ladner KJ, Chandler DS et al. miR-29 acts as a decoy in sarcomas to protect the tumor suppressor A20 mRNA from degradation by HuR. Sci Signal 2013; 6: ra63.
Ebert MS, Sharp PA . Emerging roles for natural microRNA sponges. Curr Biol 2010; 20: R858–R861.
Eiring AM, Harb JG, Neviani P, Garton C, Oaks JJ, Spizzo R et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell 2010; 140: 652–665.
Abdelmohsen K, Srikantan S, Yang X, Lal A, Kim HH, Kuwano Y et al. Ubiquitin-mediated proteolysis of HuR by heat shock. EMBO J 2009; 28: 1271–1282.
Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y et al. HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol 2000; 20: 760–769.
Ray PS, Das S . La autoantigen is required for the internal ribosome entry site-mediated translation of Coxsackievirus B3 RNA. Nucleic Acids Res 2002; 30: 4500–4508.
Acknowledgements
We acknowledge P L Fox (Cleveland Clinic), S Das (IISc Bangalore), S N Bhattacharyya (IICB, Kolkata), D Chattopadhyay (University of Calcutta) and S Sengupta (University of Calcutta) for providing reagents and laboratory facilities. We are thankful to LRI Molecular Biotechnology Core for services provided. This work was supported by a Wellcome Trust-DBT India Alliance intermediate fellowship (WT500139/Z/09/Z) to PSR, a CSIR senior research fellowship to DKP and IISER PBIP fellowship to AG.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Poria, D., Guha, A., Nandi, I. et al. RNA-binding protein HuR sequesters microRNA-21 to prevent translation repression of proinflammatory tumor suppressor gene programmed cell death 4. Oncogene 35, 1703–1715 (2016). https://doi.org/10.1038/onc.2015.235
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.235
This article is cited by
-
p53 and HuR combinatorially control the biphasic dynamics of microRNA-125b in response to genotoxic stress
Communications Biology (2023)
-
High Expression of circ_0001821 Promoted Colorectal Cancer Progression Through miR-600/ISOC1 Axis
Biochemical Genetics (2023)
-
Non-canonical features of microRNAs: paradigms emerging from cardiovascular disease
Nature Reviews Cardiology (2022)
-
The development and controversy of competitive endogenous RNA hypothesis in non-coding genes
Molecular and Cellular Biochemistry (2021)
-
Engineering combinatorial and dynamic decoders using synthetic immediate-early genes
Communications Biology (2020)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}