
Abstract
Infection by viruses having lipid-bilayer envelopes proceeds through fusion of the viral membrane with a membrane of the target cell. Viral 'fusion proteins' facilitate this process. They vary greatly in structure, but all seem to have a common mechanism of action, in which a ligand-triggered, large-scale conformational change in the fusion protein is coupled to apposition and merger of the two bilayers. We describe three examples—the influenza virus hemagglutinin, the flavivirus E protein and the vesicular stomatitis virus G protein—in some detail, to illustrate the ways in which different structures have evolved to implement this common mechanism. Fusion inhibitors can be effective antiviral agents.
Similar content being viewed by others
Main
'Enveloped' viruses—those with lipid bilayers as integral parts of their structure—enter the cells they infect by fusion of viral and host-cell membranes. One or more viral membrane proteins facilitate the various fusion steps. Several such fusion proteins have now been studied in great detail, with crystal structures determined for both the form of the protein present on the viral surface before interaction with the cell ('pre-fusion' conformation) and the form of the protein after fusion is complete ('post-fusion' conformation). The proteins show a variety of molecular architectures, but what we can infer from the various structures and from experiments both in solution and with cells suggests that all of them catalyze fusion in essentially the same way. We can even draw a rough analogy to serine proteases, which can have very different polypeptide chain folds but identical active-site mechanisms.
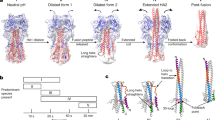
Fusion of two bilayer membranes is thermodynamically favorable, but there is a very high kinetic barrier1,2. Fusogens of all kinds lower that kinetic barrier; viral fusion proteins do so by using the free energy liberated during a protein conformational change to draw the membranes together. The general outlines of the pathway leading from two separate bilayers to a single one is relatively well understood (Fig. 1). A 'hemifusion' state—in which the apposed, proximal leaflets of the two bilayers, but not yet the distal leaflets, have merged—is almost certainly an obligatory intermediate. The structure of the hemifusion intermediate is probably stalk-like (Fig. 1d). Studies of fusion mediated by viral proteins provide some of the best evidence for hemifusion as a required intermediate stage1. There are probably substantial kinetic barriers both leading into this intermediate and leading away from it toward the product (Fig. 2).

Ambiguities remain in some aspects of this scheme (see main text). (a) The protein in the pre-fusion conformation, with its fusion peptide or loop (light green) sequestered. The representation is purely schematic, and various features of specific proteins are not incorporated—for example, the displacement of the N-terminal fragment of proteins that are cleaved from a precursor or the dimer-to-trimer rearrangement on the surface of flaviviruses. (b) Extended intermediate. The protein opens up, extending the fusion peptide or loop to interact with the target bilayer. The part of the protein that bears the fusion peptide forms a trimer cluster. (c) Collapse of the extended intermediate: a C-terminal segment of the protein folds back along the outside of the trimer core. The segments from the three subunits fold back independently, so that at any point in the process they can extend to different distances along the trimer axis, and the entire trimer can bow outward, away from the deforming membrane. (d) Hemifusion. When collapse of the intermediate has proceeded far enough to bring the two bilayers into contact, the apposed, proximal leaflets merge into a hemifusion stalk. (e) Fusion pore formation. As the hemifused bilayers open into a fusion pore, the final zipping up of the C-terminal ectodomain segments snaps the refolded trimer into its fully symmetric, post-fusion conformation, preventing the pore from resealing.

The relative heights of the various barriers are arbitrary. Fusion proteins accelerate the process by coupling traversal of these barriers to energetically favorable conformational changes.
The accumulated evidence suggests that viral fusion proteins lower the various kinetic barriers and, hence, catalyze the membrane fusion process, as follows. Step 1: The protein opens up and forms a bridge between the two bilayers (Fig. 1b). All viral fusion proteins studied so far have two membrane-interacting elements: a C-terminal transmembrane anchor that holds the protein in the viral membrane and a distinct hydrophobic patch ('fusion peptide' or 'fusion loop(s)') that ultimately interacts with the target membrane. Moreover, they all happen to be trimeric in their fusion-active state. In the initial step in the fusion reaction, the fusion protein, responding to binding of a ligand (protons in many cases, as the mechanism has evolved to respond to the low pH of an endosome3,4, but cellular or viral protein ligands in other cases), undergoes a conformational change that extends each subunit toward the target membrane and yields a contact between that membrane and the fusion peptide or loop(s) (Fig. 1a to Fig. 1b). Many fusion proteins are C-terminal fragments of a larger precursor (for example, the HA2 fragment of influenza virus hemagglutinin or the gp41 fragment of HIV Env), and initiating the fusion process requires that they must first shed their N-terminal fragment, which often contains a receptor-binding domain (for example, HA1 or gp120—see the more detailed description of influenza virus hemagglutinin below). Although strong, the evidence for an extended intermediate is indirect. The putative extended state is sometimes called a 'pre-hairpin intermediate', as the next step is collapse into a folded-back conformation. The intermediate may have a relatively long half-life; for HIV-1 gp41, the half-life seems to be many minutes, but in other cases, it may only be a few seconds. Step 2: The bridge collapses (Fig. 1c) so that the two membrane-inserted elements (the fusion peptide or loop in the target membrane and the C-terminal transmembrane anchor in the viral membrane) come together. This collapse distorts the two bilayers, probably into a nipple-like configuration, with a relatively restricted area of close approach5. Whether insertion of the fusion peptide potentiates this distortion in the target membrane, by perturbing the bilayer and lowering the distortion energy, remains uncertain. Step 3: The distortion of the individual membranes lowers the energy barrier (not necessarily symmetrically, as the anchoring of the fusion protein is different at the two ends) between separated and hemifused bilayers so that a hemifusion stalk forms (Fig. 1d). Step 4: The hemifusion stalk opens to form a transient fusion pore. A final conformational step in the protein refolding renders the open state irreversible, and the pore expands (Fig. 1e). With some fusion proteins, but not with others, the pore may flicker open and closed6,7,8. Whether flickering occurs may depend on the rapidity of the final conformational step. In most cases, steps 3 and 4 probably require concerted action of more than one fusion-protein trimer (as symbolized by the two apposed trimers in Fig. 1). The number of trimers that participate and the nature of the interaction that couples them may vary from case to case, and these issues are still matters of some debate.
To illustrate these generalizations, we describe the fusion proteins of three viruses, each with known three-dimensional structures for both the pre-fusion conformation (corresponding to Fig. 1a) and the post-fusion conformation (corresponding to Fig. 1e). These three viral fusion proteins—from influenza9, dengue10,11 and vesicular stomatitis viruses12,13—are representatives of what have come to be called class I, class II and class III viral fusion proteins, but as this typology now obscures as much as it clarifies, we avoid it here. Other viral fusion proteins for which structures in both conformational states are known include those from representative paramyxoviruses14,15 and alphaviruses16,17. The principles illustrated by those important studies reinforce the conclusions derived from the three examples chosen here.
Influenza virus hemagglutinin
The influenza virus hemagglutinin is the best characterized of all viral fusion proteins. The crystal structure of its ectodomain in a pre-fusion conformation was determined in classic work by Wiley, Wilson and Skehel in 1981 (refs. 18,19); the post-fusion conformation was finally visualized in 1994 (ref. 20), and the uncleaved precursor, HA0, in 1998 (ref. 21). The core of HA1 is a sialic acid–binding domain, borne on a stalk formed by HA2. The central feature of the stalk is a three-chain, α-helical coiled coil. HA0 and its cleaved product, pre-fusion HA1–HA2, are essentially identical in overall structure. The cleavage, which normally happens in the trans-Golgi network (TGN), but which can also occur after viral budding, leads to a modest local rearrangement, in which the newly generated N terminus of HA2 inserts into a pocket along the three-fold axis, burying the fusion peptide (the first 20–25 residues of HA2). The pocket is created by a splaying apart from each other of the C termini of the HA2 coiled-coil helices, so that the three helices diverge from the three-fold axis and from each other, rather like a narrow tripod (Fig. 3a).

(a) The pre-fusion conformation. Each subunit is shown in a different color. The binding site for the receptor, sialic acid, is at the top of each subunit, but contact with a receptor molecule is not shown. Red asterisk, the sequestered fusion peptide of the red subunit, at the N terminus of HA2. (b) HA1 dissociates from its tightly docked position in response to proton binding. Each HA1 remains flexibly tethered to the corresponding HA2 by a disulfide bond (near the bottom of the ectodomain, in the orientation shown here). (c) The extended intermediate. The loop between the shorter and longer helices in HA2 (for example, the two red helices and the loop connecting them, in b) becomes a helix, thereby translocating the fusion peptide toward the target membrane. The fusion peptides (asterisk) are shown interacting as amphipathic helices with the target bilayer. The loop-to-helix transition creates a long, three-chain coiled coil at the core of the trimer. (d) Collapse of the extended intermediate to generate the post-fusion conformation. The lower parts of the protein (as seen in the orientation in c) fold back along the outside of the three-chain coiled coil. The collapse is complete only when the two membranes have fused completely. The post-fusion conformation is shown in a 'horizontal' orientation, to correspond to the sequence in Figure 1. (e) Detail illustrating some features of the membrane-proximal region of influenza virus HA2 after fusion is complete. The N termini of the coiled-coil helices are capped by contacts with amino acid residues in the link between the fusion peptide and the coiled coil, as well as with residues near the C terminus of the ectodomain, proximal to the transmembrane helices24. This cap locks into place all the membrane-proximal components of the structure. The fusion peptides at the N termini of three HA2 chains are shown as cylinders (possible amphipathic helices) lying partly immersed in the outer leaflet of the membrane bilayer, as suggested by NMR and EPR studies27. The transmembrane segments, likely to be α-helices, are also shown as cylinders. The relationships in this drawing among the fusion peptides and the transmembrane helices, chosen to illustrate the scale of the structures and the approximate distances between them, are purely schematic, as there is no single structure yet determined experimentally that contains all the elements included here. Only the crystallographically determined components are in ribbon representation.
Sialic acid, on glycoproteins or glycolipids, is the influenza virus receptor; HA1 bears the binding site, a shallow pocket exposed on its outward-facing surface22. As the plasma membrane recycles regularly through various forms of endocytosis, the virus-receptor complex may not require a specific endocytic signal in order to reach an endosome. When the HA1:HA2 trimer encounters low pH, it undergoes a large-scale conformational rearrangement, in which HA1 separates from HA2 (Fig. 3a to Fig. 3b), except for a residual disulfide tether, and the latter effectively turns inside out20,23,24. The two key features of this HA2 refolding are a loop-to-helix transition25 in the region connecting the fusion peptide to the central coiled coil (Fig. 3b to Fig. 3c) and reorientation of the C-terminal part of the molecule so that it zips up alongside the extended coil (Fig. 3c to Fig. 3d). These correspond, respectively, to formation of the extended intermediate and to its collapse into a conformation that brings together the fusion peptide and the transmembrane anchor. The loop-to-helix transition in the N-terminal part of HA2 augments the central coiled-coil at its N-terminal end (Fig. 3c); reorientation of the C-terminal part of the protein breaks the central helices where they splay apart, at the site from which the fusion peptide has withdrawn earlier in the fusion process (Fig. 3c to Fig. 3d).
The specific sites at which protonation initiates these large-scale conformational changes may not be uniformly conserved. Mutations at widely distributed locations affect the stability of trimer interfaces that break during the rearrangement and thereby alter the threshold pH for fusion26. Conserved ionizable residues (two aspartates and a histidine) in the vicinity of the buried fusion peptide may contribute to the trigger; comparison of their interactions before and after cleavage of HA0 (and hence before and after insertion of the fusion peptide into the cavity) does not, however, lead to an obvious explanation for why HA0 does not undergo a proton-induced conformational change21. Redundant contributions to pH-regulated processes often make identification of critical residues difficult (as illustrated by the history of investigations into the hemoglobin Bohr effect) because loss of one contributing site may not lead to significant loss of fitness or to pronounced change in pH dependence.
The fusion peptide, presented to the target membrane by the loop-to-helix transition, is thought to form an amphipathic helix, by inference from its conformation on the surface of a detergent micelle and from spectroscopic data consistent with partial penetration of the outer leaflet of the lipid bilayer27. When fusion is complete, the polypeptide chain segment just C-terminal to the fusion peptide and the membrane-proximal segment of HA2 interact. The completed post-fusion conformation of HA2 includes a substructure in which residues connecting the fusion peptides to the coiled-coil and residues just N-terminal to the transmembrane anchors cap the N termini of the three central helices24 (Fig. 3e). Formation of this cap-like substructure seems to be important for the transition from hemifusion to fusion28,29.
As the extended intermediate collapses, it must bend outward, away from the nascent hemifusion stalk, to allow the two membranes to come together (Fig. 1c). The C-terminal segments of HA2 do not interact laterally with each other in the final, post-fusion conformation, and there is no reason to suppose that their zipping up is cooperative. That is, each HA2 chain can complete its refolding independently of the other two, and the loss of overall three-fold symmetry during the transition (Fig. 1c,d) is a natural consequence of this independence. Formation of the cap structure (Fig. 3e) restores global three-fold symmetry; if the transmembrane helices pass completely through the bilayer, this step probably requires the presence of an aqueous channel—that is, a committed fusion pore (Fig. 1e).
Flavivirus E
The flaviviruses (a family that includes various mosquito- and tick-borne pathogens, such as yellow fever virus, dengue virus, and tick-borne-encephalitis virus) have two envelope proteins, known as M and E. E has both receptor-binding and fusogenic activities; M is the proteolytic residuum of a precursor, prM, which is the form incorporated into immature virions. The virus assembles by budding into the endoplasmic reticulum, and furin cleavage of prM in the TGN releases most of its ectodomain. Immature virions do not fuse, even when triggered by lowering the pH, as prM is essentially a chaperone that prevents the fusion-inducing conformational transition. Cleavage of prM to M is the processing step that primes the particle for low pH–induced membrane fusion and hence is not a modification of the fusion protein itself, but rather of its companion30. The closely related alphaviruses (for example, Sindbis and Semliki Forest viruses) have a different cellular maturation pathway—they bud at the plasma membrane—but their fusion proteins (designated E1) are very similar in structure to those of the flaviviruses, and priming is likewise by cleavage of a partner protein (E2).
The mature flavivirus particle is icosahedrally symmetric and about 500 Å in diameter; its membrane bilayer has a mean diameter of about 390 Å (refs. 31,32). E covers the virion surface as an array of 90 dimers (Fig. 4a) in the pre-fusion conformation (Fig. 4b). The soluble ectodomain (sE) dimer illustrated there lacks about 50 residues connecting its C terminus with the transmembrane anchor; cryo-EM reconstructions show this so-called 'stem' region to lie in the outward-facing surface of the lipid bilayer, in a conformation that seems to be two amphipathic helices and an intervening loop31. The sE structure itself contains a central β-barrel (domain I; red in Fig. 4), from which extend two long extensions forming a distinct subdomain (domain II; yellow)10,33. The fusion loop, a short stretch at the tip of one these extensions, is buried at the dimer interface (Fig. 4a). At the C-terminal end of the sE polypeptide chain, connecting to the stem, is an immunoglobulin-like domain (domain III; blue), which probably has viral-attachment functions.

(a) The packing of 180 E subunits (90 dimers) in an icosahedral array on the surface of a flavivirus particle31. The red, yellow and blue parts of each subunit correspond respectively to domains I, II and III of the ectodomain. (b) 'Side view' of the pre-fusion, dimeric conformation of the E protein, based on the crystal structure of dengue E (residues 1–395)10, supplemented by a representation of the 'stem' segment (two helices linked by a short loop, lying in the plane of the membrane head groups) and the transmembrane anchor (a helical hairpin), derived from a cryo-EM reconstruction of the virion31. The domains in one of the two subunits are colored as in a; the other subunit is in gray. The fusion loop is at the tip of domain II, on the far right of the colored subunit, buried at the contact with domain III of the dimer partner. (c) Monomeric transition between the pre-fusion dimer and the trimeric extended intermediate. The three subunits that will associate into the extended intermediate in d are not yet in contact. The drawing embodies the suggestion that domains I and II have swung outward, while domain III and the stem remain oriented against the membrane roughly as in the pre-fusion state. The fusion loop is now at the top of the diagram and is shown already interacting with the target bilayer. (d) Extended intermediate. Domains I and II have associated into the trimeric core of the post-fusion conformation, but domain III has not yet flipped over (upper arrows) to dock against them47. To indicate that the stem segment must then zip back along the trimer core (lower arrows), the stem is represented by loops 'poised' to reconfigure. (e) Post-fusion conformation. Domain III has reoriented, and the stem (dashed line, as there is no direct structural information on its conformation or exact position in the post-fusion trimer) connects it to the transmembrane anchor, now brought together with the fusion loop in the single, fused bilayer. The post-fusion conformation is shown in a 'horizontal' orientation, to correspond to the sequence in Figure 1.
The response to lowered pH in the presence of a membrane leads to the following sequence of molecular events11,34,35. The dimers dissociate, allowing the individual subunits to swing outward36. The now-exposed fusion loops insert into the target membrane, facilitating reclustering of the subunits into trimers. Collapse of the extended, trimeric intermediate that results from these events can then proceed, by rotation of domain III in each subunit about the segment that links it to domain I and (presumably) by zipping up of the stem alongside the clustered domains II. Protonation of one or two conserved histidine residues at the domain I–domain III interface probably contributes to initiating this process37. Mutations around a hydrophobic pocket at the domain I–domain II interface also affect the pH threshold; this region undergoes a hinge-like change during the transition, and alterations in the bulk of the hydrophobic side chains that face the pocket could influence both the kinetic barrier and the net free energy change of the process10.
The structures of E protein trimers from dengue and TBE viruses show the C terminus of the ectodomain projecting toward the fusion loop; the stem segment has not yet been detected directly in a crystal structure, but as the transmembrane segment at its C terminus must reside after fusion in the same membrane as the fusion loop, the stem must in some sense zip up alongside the clustered domains II (refs. 10,35) (Fig. 4d,e). The absence of a structure that includes the full stem also leaves open the question of whether a defined 'cap' completes the zipping up. The residues just N-terminal to the transmembrane segment are mostly hydrophobic, and one can imagine that they form some sort of tight interaction with the fusion loop or its rim and perhaps also with the lipid bilayer itself.
Vesicular stomatitis virus G
Unlike the low pH–induced conformational change in the fusion proteins just described, the shift between high- and low-pH forms of the glycoprotein (G) of rhabdoviruses (for example, vesicular stomatitis virus (VSV) and rabies virus) seems to be reversible38. That is, virions inactivated by prolonged incubation at pH < 6 can be reactivated by raising the pH to neutral or above, and both conformations of the trimeric protein described here can be obtained from the same protein preparation.
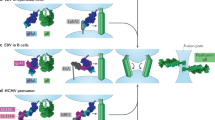
The structures of VSV-G in high-pH (pre-fusion)13 and low-pH (presumably post-fusion)12 conformations show that, despite these distinctive properties, the fundamental characteristics of the protein and of how it facilitates fusion are similar to those of influenza virus hemagglutinin or flavivirus E (Fig. 5). G has two hydrophobic loops that can cross-link to derivatized membrane lipids39; the structures show that these loops, each of which links a pair of antiparallel β-strands, lie next to each other at the tip of an elongated domain (Fig. 5a). In the pre-fusion conformation, these domains face the viral membrane. In the post-fusion conformation, they cluster around the three-fold axis, presenting the fusion loops to the target membrane (Fig. 5e). The connectivity of the strands joined by the fusion loops is different from the connectivity in domain II of flavivirus E and alphavirus E1 (that is, the domains themselves have different folds), but the general picture is quite similar: hydrophobic residues (including at least one tryptophan) are displayed on tightly structured loops at the end of an elongated domain.

(a) Pre-fusion trimer. The three subunits are in red, blue and green. The fusion loops (asterisk) are held away from the target membrane. The crystal structure13 does not include about 40 residues, represented here for each subunit by a slightly wavy line, that connect to the transmembrane anchor. (b) Pre-fusion conformation of one subunit (in the orientation of the red subunit in a). Core domain, red; α-helix at the trimer contact, light blue; two-part fusion apparatus, dark blue; C-terminal segment, dark green. Dashed line, the part of the C-terminal segment that is missing from the crystal structure; letter N, the N terminus. (c) Suggested extended intermediate conformation of one subunit, colored as in b. The fusion domains have reoriented (curved arrow in b), with the fusion loops (asterisk) now in contact with the target membrane; the reorientation seems to be driven in part by a loop-to-helix transition that elongates the helix at the trimer contact. The C-terminal segment still connects to the viral membrane (dashed arrow), but it must fold back along the outside of the trimer (curved arrow) to complete the transition to the post-fusion conformation. (d) Post-fusion conformation of one subunit, in the orientation and colors of the subunit in b and c. The C-terminal segment has folded back, and it now projects toward the fusion loops. (e) Post-fusion conformation of the trimer12, with colors as in a. It is shown in a 'horizontal' orientation, to correspond to the sequence in Figure 1.
The rhabdovirus G protein has a more intricately folded structure than do flavivirus E and alphavirus E1. We can analyze the structure as three domains, each of roughly invariant fold, linked by segments that change conformation as the domains rotate with respect to one another (Fig. 5). A core domain, based on a β-sandwich (red in Figs. 5b–d) and a relatively long α-helix (light blue in Figs. 5b–d), dominates the three-fold contact in the high-pH (pre-fusion) conformation. This domain contains residues from the N-terminal segment of the polypeptide chain and residues from near the C-terminal part of the chain: we can consider it a framework around which the rest of the molecule reorients. The other two domains form a jointed, two-part fusion machinery. The net effect of their rotations relative to each other and to the core domain is to translate the fusion loops, at the tip of the outermost domain, away from the viral membrane and toward the target membrane. In a likely extended intermediate conformation (shown in Fig. 5c, but for which there are as yet no direct structural data), the C-terminal segment still connects 'downward', even as the fusion loops interact with the target. In the fully rearranged, low-pH conformation (Figs. 5d,e), the C-terminal segment has zipped up along the fusion domains, much as in the flavivirus fusion transition. Aside from this zipping up of the C terminus, the most pronounced conformational change is the loop-to-helix transition of the 12-residue segment between the fusion domains and the core domain (Fig. 5b to Fig. 5c, blue helix), reminiscent of the even more notable loop-to-helix transition in influenza HA2. As in hemagglutinin, the loop-to-helix transition in VSV-G creates a long, three-chain coiled coil at the trimer axis and seems to propel the fusion loops toward the target membrane.
The herpesvirus fusion protein, gB, is a 'stretched' version of VSV-G40. This unexpected similarity between fusion proteins of a DNA virus and a negative-strand RNA virus has led to some evolutionary speculations, but the important result from the point of view of understanding fusion mechanisms is that information about one protein (for example, the identification of the rhabdovirus fusion loops) can be carried over to the other41. Only the presumptive post-fusion structure of gB has been determined so far. The gB conformational transition is triggered not by changes in pH, but rather by receptor binding to another surface protein, gD. How a binding-induced conformational change in gD leads to the reorganization of gB remains to be worked out. The role of one further, conserved, herpesvirus surface protein, the gH–gL heterodimer, is likewise still undetermined.
Activating and initiating
For most fusion proteins, we can distinguish a priming step and an activating or triggering step for the sequence of events that follows. Priming is usually the result of proteolysis—either of the fusion protein itself, as in the case of influenza hemagglutinin or retroviral Env, or of an accompanying protein, as in the case of flavi- and alphaviruses. For these viruses, priming occurs during transport of the immature glycoprotein to the cell surface, either before assembly of the virus particle by budding at the cell surface or after a formation of an immature particle by budding through an internal membrane. The glycoproteins of other viruses—Ebola virus and severe acute respiratory syndrome (SARS)coronavirus in particular—require cleavage by endosomal cathepsins B or L during cell entry rather than during maturation42,43. The SARS coronavirus spike protein, S, is a trimer of uncleaved chains on the virion surface, but receptor binding seems to make it susceptible to cathepsin L (ref. 43). In addition to making the fusogenic conformational rearrangement possible, cathepsin attack, by releasing a covalent constraint, may also be sufficient to induce the rearrangement—that is, cleavage may be a triggering as well as a priming step, after a 'pre-priming' by the receptor interaction. The Ebola virus glycoprotein, GP0, is cleaved by furin to GP1 and GP2 before incorporation into virions. Degradation of GP1 by cathepsins may be part of the triggering step, to release GP2 from the constraints that prevent its fusogenic conformational rearrangement, but some further endosomal activity seems to be required as well44.
A primed fusion protein is metastable in its pre-fusion state. A covalent peptide bond (either in the fusion protein itself, as in hemagglutinin, or in the chaperone or guard protein, as in dengue prM-E) restrains the initial, folded conformation of the precursor. Once that covalent restraint has gone (irreversibly), a high kinetic barrier still separates the primed from the post-fusion conformation. The trigger that lowers this barrier (or provides the required activation energy) can be binding of a proton, for viruses that have evolved to detect a low-pH, endosomal environment, or binding of a co-receptor in some other cases (for example, HIV-1); for herpesviruses and paramyxoviruses, the trigger is an altered lateral contact with another viral surface protein that has itself changed conformation because of binding with a cellular receptor. Whatever the trigger, association with the ligand alters the free energy profile, so that rearrangement to the post-fusion state is rapid. The free energy liberated by the rearrangement can then be used to overcome the barrier to merging two membrane bilayers.
The initial response to the trigger is probably formation of an extended intermediate. For influenza virus hemagglutinin, the first event must be loosening of the restraints on HA2 imposed by HA1. HA1 is linked to HA2 by a disulfide near the base of the trimer, so that it cannot dissociate completely, but it clearly must get out of the way of the loop-to-helix transition and the subsequent zippering step45 (Fig. 3b). For flavivirus E proteins, the initial dissociation of the dimer probably allows the monomer to flex outward, encounter the target membrane and associate into trimers36 (Fig. 4b–d). Soluble E ectodomain dimers dissociate reversibly at low pH and redimerize if the pH is returned to neutral. If liposomes are present in the low-pH step, however, the fusion loops insert into the lipid bilayer and the protein trimerizes irreversibly46. The bilayer is a surface catalyst in this context: by aligning the individual subunits in a preferred orientation (fusion-loop tip in the membrane), it lowers the barrier that separates the free monomer from the true minimum-energy state represented by the post-fusion trimer.
The case of VSV-G is puzzling. If the protein could undergo the complete transition illustrated in Figure 5, then it should become inactivated at low pH, by inverting during the zipping-up process so that its fusion loops ultimately insert into the viral membrane. The hemagglutinins of many influenza strains undergo just this kind of inactivation when the pH is lowered in the absence of attachment to a target membrane. One possibility is that on the viral surface, the protein can shift reversibly between the states represented by Figure 5b and Figure 5c, but insertion of the fusion loops into a target membrane somehow favors the further (irreversible?) transition to the state in Figure 5d.
The extended intermediate
The postulated extended intermediate has been characterized functionally for HIV-1 gp41 and somewhat less extensively for flavi- and alphaviruses47 and paramyxoviruses48. The post-fusion conformation of the gp41 ectodomain is particularly simple—just a trimer of hairpins, in which both prongs of the hairpin are α-helices49,50,51(Fig. 6). Approximately 50 residues immediately C-terminal to the fusion peptide (designated HR1, where “HR” stands for “heptad repeats”) form a central, three-chain coiled coil. A loop that contains a conserved disulfide bond connects the HR1 segment to a second heptad-repeat element, HR2, which forms an outer-layer α-helix. Peptides from this outer layer can inhibit the fusion process52,53. The mechanism involves association of the peptide with the inner core, preventing transition to the post-fusion conformation. The lifetime of the intermediate, as detected by the capacity of such peptides to inhibit fusion during the period after attachment and initiation of conformation change, is at least several minutes54; its magnitude is probably determined by the resistance of the two membranes to being pulled toward each other. Without that resistance, the zipping up of the outer layer might be too rapid for peptide to intervene. HIV fusion occurs at the cell surface, and one such inhibitory peptide (T-20, or enfuvirtide) is a clinically useful drug55. Mutations conferring resistance to T-20 can occur at various positions in the envelope protein, including residues in gp120 (ref. 56). Some of the mutations in HR1 that reduce T-20 binding also retard fusion and enhance sensitivity to antibodies targeting the membrane-proximal region of gp41 (ref. 57). There is also biochemical evidence that the extended intermediate is the target for this set of neutralizing antibodies58.

In the extended intermediate (left), the HR1 segment of each of the three subunits is shown as an α-helix, and the HR2 segment as an extended chain. The fusion peptides are imagined to be inserted into or against the target-cell membrane (top) and the transmembrane anchors pass into the viral membrane (bottom). In the post-fusion conformation (right), the HR2 segment has zipped up into a helix along the outside of the HR1 three-chain coiled-coil, creating a 'trimer of hairpins', and the two membranes have fused. Courtesy Gaël McGill (see http://www.molecularmovies.org).
Fusion of flavi- and alphaviruses occurs within endosomes, but it can be induced experimentally at the cell surface by exposing receptor-bound virus to a brief pulse of low-pH medium. Soluble domain III of the fusion protein, or domain III plus the stem, can inhibit fusion when present during acidification47. The target of inhibition is presumably the extended intermediate, in which domain III and the stem have yet to curl around and project back toward the fusion loops at the tip of domain II.
Four questions
How many fusion-protein trimers contribute to formation of a fusion pore? Diagrams such as those in Figure 1 implicitly suggest participation of more than one fusion-protein trimer per fusion event. But there seems to be no fundamental reason why the fusion machinery needs to surround the hemifusion stalk or fusion pore. The energy barrier that must be overcome en route to a hemifusion stalk is thought to be about ∼40–50 kcal mol−1 (refs. 2,5). A free energy of roughly this magnitude could in principle be recovered from the collapse of just one or two trimers, if the interactions driving refolding were strong enough.
Cell-cell fusion mediated by influenza virus hemagglutinin responds nonlinearly to the concentration of hemagglutinin on the cell surface, in a manner consistent with cooperativity in fusion-pore formation59. An estimate obtained from analysis of the lag time to fusion (after a drop in pH) for red blood cells with hemagglutinin-expressing cells suggests that three or four hemagglutinin trimers might participate in a single fusion event. As the resistance of the two membranes to deformation and the height of the kinetic barrier leading to hemifusion will probably change with membrane composition, it is likely that the actual number of hemagglutinin trimers required will vary from cell type to cell type.
Fusion of two cells is a complex event, potentially involving a number of distinct pores at different positions across the area of contact. Experiments with HIV and HIV pseudotypes containing a mixture of cleavable and uncleavable (and hence active and inactive) fusion proteins suggest that only one active retroviral envelope trimer is sufficient for fusion, while confirming the requirement for several hemagglutinin trimers60. The fusion proteins of HIV and other retroviruses may have evolved to manage with a single fusion protein, as the complement of trimers on the virion surface can be rather sparse: estimates of 15–20 trimers total have been made for HIV. Rough calculations based on the known (Kd ≈ 1 nM) affinity of an HIV gp41 outer-layer peptide for a trimeric inner core suggest that the free energy liberated during collapse of the extended intermediate to the folded-back trimer of hairpins might approach the estimate of ∼40–50 kcal mol−1 for the hemifusion barrier.
Cooperativity, if it occurs, could have two distinct mechanisms. One requires lateral contacts between adjacent trimers in a ring around the fusion pore site. A specific structure for such a ring has been proposed for the alphavirus fusion protein17. Lateral contacts would couple the conformational change of one trimer to that in another, as in allosteric regulation of multi-subunit enzymes. The other mechanism, which will contribute even if the former does also, results simply from the response of the two membranes to the distortions necessary to promote fusion. At least two properties of bilayer membranes will cause them to resist the collapse of the extended protein intermediates that bridge them. One is the energy of bending—for example, into the nipple-like configuration (Fig. 1c); the other is the so-called 'hydration force', which creates a substantial barrier when apposing membranes come closer than about 10–20 Å (ref. 61). Because all the proteins participating in a fusion event bridge the same pair of membranes, the behavior of one extended intermediate is not independent of the behavior of a neighboring one—they are coupled by the deformation energies of the two bilayers they connect.
Does insertion of the fusion peptide or loop(s) into the target membrane perturb the bilayer in a way that lowers the kinetic barrier for hemifusion, or does collapse of the extended intermediate do most of the work? Deformation of a planar membrane into a nipple-like bud (Fig. 1c) creates a nearly hemispherical cap, with roughly uniform positive curvature, and a flared region joining it to the planar membrane of the membrane. The flared region has positive curvature in one direction (around the axis of the nipple) and negative in the other (within the plane of the cross-section in Fig. 1c), and hence little net elastic distortion. Using reasonable dimensions for the bud5, one can estimate the difference in area between the inner and outer leaflets of the positively curved cap as 2 × 104 Å2. Available structural data show that fusion loops and fusion peptides insert only partway into the outer leaflet of the target membrane, displacing lipid head groups laterally and therefore favoring curvature of the leaflet11,17,27. But their contribution is probably only a modest fraction of the total curvature in the cap. For example, the tip of one flavivirus trimer occupies about 800 Å2. Hence, insertion of the tips of even five or six trimers will provide only 20%–25% of the required distortion. The direction of curvature promoted by the observed insertion does suggest, however, that as the extended intermediates collapse, their fusion peptides or fusion loops may tend to concentrate in the positively curved cap and migrate toward the developing hemifusion stalk. At this stage, they may contribute substantially to the difference between the areas of the inner and outer leaflets.
Does the membrane-proximal segment of the ectodomain have a mechanistic role? The membrane-proximal segments (10–15 residues) of many fusion-protein ectodomains have characteristic hydrophobic and often relatively tryptophan-rich sequences. At the end of the fusion-inducing conformational change, these segments are apposed to the merged membrane and could in principle interact with the membrane or the fusion peptides/loops (or both). The membrane-proximal segments of influenza virus hemagglutinin might have a defined role in stabilizing the final, post-fusion structure (and hence potentially in stabilizing an open fusion pore) (Fig. 3e). The membrane-proximal region of HIV gp41 has been studied with particular attention, as it is the target of two well-characterized, broadly neutralizing antibodies, both of which seem to target the extended intermediate rather than the pre-fusion envelope trimer. Some recent NMR experiments suggest that it could form a bent amphipathic helix lying in the outer part of the membrane bilayer62.
What structural rearrangements lower the kinetic barrier between hemifusion and fusion-pore formation? If influenza virus hemagglutinin is linked to a glycosyl phosphatidylinositol anchor or to a truncated protein anchor that does not completely traverse the membrane, the fusion process halts at hemifusion and proceeds forward very slowly63,64,65. It thus seems that collapse of the extended intermediate—the transition that induces hemifusion-stalk formation—is not sufficient to drive the fusion process to completion. At the hemifusion stage, a hemagglutinin-induced fusion pore can flicker open and closed6, and we may infer that some property of the fusion protein, connected with complete traversal of the membrane, must accelerate the forward reaction. The capping of the central helix in influenza HA2 (Fig. 3e) shows that formation of a well-defined structure, with tight hydrophobic and hydrogen-bonding interactions among the participating amino acid residues, brings about a concerted closure of the conformational transition, drawing the near ends of the transmembrane helices together and causing the cytoplasmic segments on the opposite side of the membrane to pass into or through the nascent fusion pore24. Presence of these segments in the aqueous channel of a fusion pore will prevent its resealing and may explain how a cytoplasmic element at the far end of the transmembrane anchor can drive the fusion process forward65. In other words, formation of a cap on the HA2 helical bundle when zipping-up is complete becomes a mechanism for making pore formation irreversible28 because cap formation couples to a translocation, into or through the pore, of material from the cytoplasmic face of the bilayer.
Fusion inhibitors
Ligands can retard or block viral entry if they bind selectively to any conformation of the fusion protein that precedes, in the fusion pathway, merger of the two bilayers. For example, inhibition of fusion is a mechanism by which some neutralizing antibodies block viral infection. T-20, the outer-layer peptide from HIV gp41 described above, demonstrates that agents selected or designed to inhibit the fusion-inducing conformational change can be useful in the clinic55. Small molecules have been found that block HIV fusion, by screening for compounds that compete with an outer-layer peptide for binding to the three-helix bundle of the post-fusion gp41 inner core66. It might be possible to extend this strategy to many other viruses, as zipping up of an outer-layer segment against the inner core of an intermediate is a universal feature of the fusion-inducing conformational changes illustrated in Figures 3, 4, 5, 6.
Small molecules have also been found that bind HIV gp120 in its pre-fusion conformation, thereby raising the barrier to the initial steps of the fusion sequence67. The site for these molecules is probably a pocket in gp120 that closes up in the initial, CD4-induced conformational transition68. Productive receptor binding must expel the compound from this site and thus incur an inhibitor-specific free-energy cost. A candidate has been described for a similar kind of site on dengue virus E—a pocket that disappears when the protein starts to rearrange10. Indeed, any protein that undergoes a large-scale conformational change is likely to have some sort of small-molecule binding site that changes or disappears in the transition from one state to the other. We need now to understand when it is practical to exploit this property for discovery of useful compounds. A structure-based, mechanistic analysis of membrane fusion, as outlined in this review, can clearly contribute to the discovery process.
References
Chernomordik, L.V. & Kozlov, M.M. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72, 175–207 (2003).
Chernomordik, L.V., Zimmerberg, J. & Kozlov, M.M. Membranes of the world unite! J. Cell Biol. 175, 201–207 (2006).
Helenius, A., Kartenbeck, J., Simons, K. & Fries, E. On the entry of Semliki Forest virus into BHK-21 cells. J. Cell Biol. 84, 404–420 (1980). One of several papers in which Helenius, Simons and their co-workers showed that the acidic pH of an endosome is a trigger for viral fusion. The demonstration that viruses have evolved to 'sense' the local proton concentration was a contribution both to our knowledge of viral entry mechanisms and to our understanding of the properties of endocytic pathways more generally.
White, J. & Helenius, A. pH-dependent fusion between Semliki Forest virus membrane and liposomes. Proc. Natl. Acad. Sci. USA 77, 3273–3277 (1980).
Kuzmin, P.I., Zimmerberg, J., Chizmadzhev, Y.A. & Cohen, F.S. A quantitative model for membrane fusion based on low-energy intermediates. Proc. Natl. Acad. Sci. USA 98, 7235–7240 (2001).
Zimmerberg, J., Blumenthal, R., Sarkar, D.P., Curran, M. & Morris, S.J. Restricted movement of lipid and aqueous dyes through pores formed by influenza hemagglutinin during cell fusion. J. Cell Biol. 127, 1885–1894 (1994).
Plonsky, I. & Zimmerberg, J. The initial fusion pore induced by baculovirus GP64 is large and forms quickly. J. Cell Biol. 135, 1831–1839 (1996).
Melikyan, G.B., Markosyan, R.M., Brener, S.A., Rozenberg, Y. & Cohen, F.S. Role of the cytoplasmic tail of ecotropic Moloney murine leukemia virus Env protein in fusion pore formation. J. Virol. 74, 447–455 (2000).
Skehel, J.J. & Wiley, D.C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 69, 531–569 (2000).
Modis, Y., Ogata, S., Clements, D. & Harrison, S.C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 100, 6986–6991 (2003).
Modis, Y., Ogata, S., Clements, D. & Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 427, 313–319 (2004).
Roche, S., Bressanelli, S., Rey, F.A. & Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313, 187–191 (2006).
Roche, S., Rey, F.A., Gaudin, Y. & Bressanelli, S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315, 843–848 (2007).
Yin, H.S., Paterson, R.G., Wen, X., Lamb, R.A. & Jardetzky, T.S. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA 102, 9288–9293 (2005).
Yin, H.S., Wen, X., Paterson, R.G., Lamb, R.A. & Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439, 38–44 (2006).
Lescar, J. et al. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105, 137–148 (2001).
Gibbons, D.L. et al. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427, 320–325 (2004).
Wilson, I.A., Skehel, J.J. & Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 289, 366–373 (1981). The ground-breaking initial structural result of the Skehel-Wiley collaboration on the influenza virus hemagglutinin. A milestone in structural biology and surface-glycoprotein biochemistry, this paper antedated by nearly 15 years the next report of a distinct viral fusion-protein structure. It helped shape the entire field of enveloped virus entry and viral antigenicity.
Wiley, D.C., Wilson, I.A. & Skehel, J.J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289, 373–378 (1981).
Bullough, P.A., Hughson, F.M., Skehel, J.J. & Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371, 37–43 (1994). This paper, which reports the structure of the post-fusion conformation of influenza virus HA2, capped a twelve-year effort to visualize the product of the conformational transition discovered by Skehel et al.23. The extent of the HA2 refolding was unanticipated, and seeing it changed our appreciation of the likely repertoire of protein conformational transitions.
Chen, J. et al. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95, 409–417 (1998).
Weis, W. et al. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 333, 426–431 (1988).
Skehel, J.J. et al. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc. Natl. Acad. Sci. USA 79, 968–972 (1982). The discovery that proton binding (low pH) triggers a profound conformational change in influenza virus hemagglutinin. The authors were able to infer the essential characteristics (although not yet the extent) of the conformational change in hemagglutinin, by insightful use of selective proteolysis and by thoughtful interpretation of solubilizing effects of nonionic detergents.
Chen, J., Skehel, J.J. & Wiley, D.C. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA2 subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA 96, 8967–8972 (1999).
Carr, C.M. & Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73, 823–832 (1993).
Daniels, R.S. et al. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40, 431–439 (1985).
Han, X., Bushweller, J.H., Cafiso, D.S. & Tamm, L.K. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 8, 715–720 (2001).
Borrego-Diaz, E., Peeples, M.E., Markosyan, R.M., Melikyan, G.B. & Cohen, F.S. Completion of trimeric hairpin formation of influenza virus hemagglutinin promotes fusion pore opening and enlargement. Virology 316, 234–244 (2003).
Park, H.E., Gruenke, J.A. & White, J.M. Leash in the groove mechanism of membrane fusion. Nat. Struct. Biol. 10, 1048–1053 (2003).
Heinz, F.X. et al. Structural changes and functional control of the tick-borne encephalitis virus glycoprotein E by the heterodimeric association with protein prM. Virology 198, 109–117 (1994).
Zhang, W. et al. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat. Struct. Biol. 10, 907–912 (2003).
Mukhopadhyay, S., Kim, B.S., Chipman, P.R., Rossmann, M.G. & Kuhn, R.J. Structure of West Nile virus. Science 302, 248 (2003).
Rey, F.A., Heinz, F.X., Mandl, C., Kunz, C. & Harrison, S.C. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 375, 291–298 (1995).
Allison, S.L. et al. Oligomeric rearrangement of tick-borne encephalitis virus envelope proteins induced by an acidic pH. J. Virol. 69, 695–700 (1995).
Bressanelli, S. et al. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 23, 728–738 (2004).
Stiasny, K., Kossl, C., Lepault, J., Rey, F.A. & Heinz, F.X. Characterization of a structural intermediate of flavivirus membrane fusion. PLoS Pathog. 3, e20 (2007).
Kampmann, T., Mueller, D.S., Mark, A.E., Young, P.R. & Kobe, B. The role of histidine residues in low-pH-mediated viral membrane fusion. Structure 14, 1481–1487 (2006).
Roche, S. & Gaudin, Y. Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology 297, 128–135 (2002).
Durrer, P., Gaudin, Y., Ruigrok, R.W., Graf, R. & Brunner, J. Photolabeling identifies a putative fusion domain in the envelope glycoprotein of rabies and vesicular stomatitis viruses. J. Biol. Chem. 270, 17575–17581 (1995).
Heldwein, E.E. et al. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313, 217–220 (2006).
Hannah, B.P., Heldwein, E.E., Bender, F.C., Cohen, G.H. & Eisenberg, R.J. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 81, 4858–4865 (2007).
Chandran, K., Sullivan, N.J., Felbor, U., Whelan, S.P. & Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308, 1643–1645 (2005).
Simmons, G. et al. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 102, 11876–11881 (2005).
Schornberg, K. et al. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 80, 4174–4178 (2006).
Godley, L. et al. Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell 68, 635–645 (1992).
Stiasny, K., Allison, S.L., Schalich, J. & Heinz, F.X. Membrane interactions of the tick-borne encephalitis virus fusion protein E at low pH. J. Virol. 76, 3784–3790 (2002).
Liao, M. & Kielian, M. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus membrane fusion. J. Cell Biol. 171, 111–120 (2005).
Russell, C.J., Jardetzky, T.S. & Lamb, R.A. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20, 4024–4034 (2001).
Blacklow, S.C., Lu, M. & Kim, P.S. A trimeric subdomain of the simian immunodeficiency virus envelope glycoprotein. Biochemistry 34, 14955–14962 (1995).
Chan, D.C., Fass, D., Berger, J.M. & Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89, 263–273 (1997).
Weissenhorn, W., Dessen, A., Harrison, S.C., Skehel, J.J. & Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387, 426–430 (1997).
Wild, C.T., Shugars, D.C., Greenwell, T.K., McDanal, C.B. & Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 91, 9770–9774 (1994).
Furuta, R.A., Wild, C.T., Weng, Y. & Weiss, C.D. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 5, 276–279 (1998).
Munoz-Barroso, I., Durell, S., Sakaguchi, K., Appella, E. & Blumenthal, R. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J. Cell Biol. 140, 315–323 (1998).
Kilby, J.M. & Eron, J.J. Novel therapies based on mechanisms of HIV-1 cell entry. N. Engl. J. Med. 348, 2228–2238 (2003).
Reeves, J.D. et al. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. USA 99, 16249–16254 (2002).
Reeves, J.D. et al. Enfuvirtide resistance mutations: impact on human immunodeficiency virus envelope function, entry inhibitor sensitivity, and virus neutralization. J. Virol. 79, 4991–4999 (2005).
Frey, G. et al. A fusion-intermediate state of HIV-1 gp41 targeted by broadly neutralizing antibodies. Proc. Natl. Acad. Sci. USA 105, 3739–3744 (2008).
Danieli, T., Pelletier, S.L., Henis, Y.I. & White, J.M. Membrane fusion mediated by the influenza virus hemagglutinin requires the concerted action of at least three hemagglutinin trimers. J. Cell Biol. 133, 559–569 (1996).
Yang, X., Kurteva, S., Ren, X., Lee, S. & Sodroski, J. Subunit stoichiometry of human immunodeficiency virus type 1 envelope glycoprotein trimers during virus entry into host cells. J. Virol. 80, 4388–4395 (2006).
Rand, R.P. & Parsegian, V.A. Physical force considerations in model and biological membranes. Can. J. Biochem. Cell Biol. 62, 752–759 (1984).
Sun, Z.Y. et al. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity 28, 52–63 (2008).
Kemble, G.W., Danieli, T. & White, J.M. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 76, 383–391 (1994).
Melikyan, G.B., White, J.M. & Cohen, F.S. GPI-anchored influenza hemagglutinin induces hemifusion to both red blood cell and planar bilayer membranes. J. Cell Biol. 131, 679–691 (1995).
Armstrong, R.T., Kushnir, A.S. & White, J.M. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J. Cell Biol. 151, 425–437 (2000).
Frey, G. et al. Small molecules that bind the inner core of gp41 and inhibit HIV envelope-mediated fusion. Proc. Natl. Acad. Sci. USA 103, 13938–13943 (2006).
Lin, P.F. et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl. Acad. Sci. USA 100, 11013–11018 (2003).
Chen, B. et al. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433, 834–841 (2005).
Acknowledgements
I have benefited from discussions (at various times, not all in direct connection with this review) with J. Skehel, F. Heinz, F. Rey, M. Kielian, J. White, J. Zimmerberg and B. Chen, but each will probably dissent from one or more of my interpretations or from my choice of examples and selection of references. I acknowledge support related to the topic of this review from US National Institutes of Health grants AI-57159 and CA-13202, and from the Howard in the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Harrison, S. Viral membrane fusion. Nat Struct Mol Biol 15, 690–698 (2008). https://doi.org/10.1038/nsmb.1456
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.1456
This article is cited by
-
Exosomal transmission of viruses, a two-edged biological sword
Cell Communication and Signaling (2023)
-
Cell entry and release of quasi-enveloped human hepatitis viruses
Nature Reviews Microbiology (2023)
-
Structural characterisation of hemagglutinin from seven Influenza A H1N1 strains reveal diversity in the C05 antibody recognition site
Scientific Reports (2023)
-
Activation energy and force fields during topological transitions of fluid lipid vesicles
Communications Physics (2022)
-
Recent Developments in Single-Virus Fusion Assay
The Journal of Membrane Biology (2022)
