
Abstract
Increasing evidence suggests that reductions in brain-derived neurotrophic factor (BDNF) and its receptor tyrosine receptor kinase B (TrkB) may have a role in the pathogenesis of Alzheimer's disease (AD). However, the efficacy and safety profile of BDNF therapy (eg, gene delivery) remains to be established toward clinical trials. Here, we evaluated the effects of 7,8-dihydroxyflavone (7,8-DHF), a recently identified small-molecule TrkB agonist that can pass the blood–brain barrier, in the 5XFAD transgenic mouse model of AD. 5XFAD mice at 12–15 months of age and non-transgenic littermate controls received systemic administration of 7,8-DHF (5 mg/kg, i.p.) once daily for 10 consecutive days. We found that 7,8-DHF rescued memory deficits of 5XFAD mice in the spontaneous alternation Y-maze task. 5XFAD mice showed impairments in the hippocampal BDNF-TrkB pathway, as evidenced by significant reductions in BDNF, TrkB receptors, and phosphorylated TrkB. 7,8-DHF restored deficient TrkB signaling in 5XFAD mice without affecting endogenous BDNF levels. Meanwhile, 5XFAD mice exhibited elevations in the β-secretase enzyme (BACE1) that initiates amyloid-β (Aβ) generation, as observed in sporadic AD. Interestingly, 7,8-DHF blocked BACE1 elevations and lowered levels of the β-secretase-cleaved C-terminal fragment of amyloid precursor protein (C99), Aβ40, and Aβ42 in 5XFAD mouse brains. Furthermore, BACE1 expression was decreased by 7,8-DHF in wild-type mice, suggesting that BDNF-TrkB signaling is also important for downregulating baseline levels of BACE1. Together, our findings indicate that TrkB activation with systemic 7,8-DHF can ameliorate AD-associated memory deficits, which may be, at least in part, attributable to reductions in BACE1 expression and β-amyloidogenesis.
Similar content being viewed by others


INTRODUCTION
Levels of brain-derived neurotrophic factor (BDNF) and its receptor tyrosine receptor kinase B (TrkB) are decreased in the hippocampus and cerebral cortex early in the progression of Alzheimer's disease (AD) (Hu and Russek, 2008; Schindowski et al, 2008; Tapia-Arancibia et al, 2008; Zuccato and Cattaneo, 2009; Ginsberg et al, 2010). As BDNF-TrkB signaling is crucial for neuronal plasticity (Minichiello, 2009; Pang and Lu, 2004), it is hypothesized that deficient BDNF-TrkB pathways may underlie amyloid-β (Aβ)-induced neurotoxicity, synaptic dysfunction, and memory deficits in this devastating neurodegenerative disorder. This hypothesis is supported by recent findings that BDNF administration or gene delivery to in vivo AD models such as amyloid precursor protein (APP) transgenic mice produces beneficial effects including improvements in learning and memory (Arancibia et al, 2008; Nagahara et al, 2009). Furthermore, neural stem cell transplantation is shown to rescue impaired spatial learning and memory of triple transgenic (3xTg-AD) mice in the Morris water maze probably by elevating hippocampal BDNF levels (Blurton-Jones et al, 2009). However, molecular mechanisms, by which BDNF-TrkB deficiency may contribute to the pathogenesis of AD and its restoration may counteract AD-related phenotypes, are poorly understood. Moreover, although these gain-of-function experiments suggest the possibility of developing BDNF therapy for AD treatments, the biomedical approaches used need to clear several hurdles to be translated well into clinical testing. For example, clinical trials with recombinant BDNF have not been successful presumably because of a short in vivo half-life or limited diffusion, while much work remains for establishing the safety and methodology to provide regulated release of BDNF in gene delivery and neural stem cell strategies (Zuccato and Cattaneo, 2009).
Recent screening of a chemical library has identified a flavone derivative 7,8-dihydroxyflavone (7,8-DHF) as the first small-molecule compound that binds with high affinity and specificity to the TrkB receptor, provokes its dimerization and autophosphorylation, and activates its downstream signaling cascade (Jang et al, 2010; Liu et al, 2010). Remarkably, systemic administration of 7,8-DHF can activate TrkB receptors in brains as evidenced by increases in phosphorylated TrkB, and induce BDNF-like behavioral phenotypes such as enhanced learning and memory, and antistress or antidepressant-like effects in rodents (Andero et al, 2010; Andero et al, 2011; Choi et al, 2010; Jang et al, 2010; Liu et al, 2010). Therefore, the small-molecule TrkB receptor agonist 7,8-DHF represents a novel and useful tool to investigate the role of BDNF-TrkB signaling in vivo. However, it has not been examined whether this compound may exert beneficial effects against AD-related pathology or neuronal dysfunction.
In this study, we performed preclinical evaluation of the TrkB agonist 7,8-DHF in a mouse model of AD. We used 5XFAD transgenic mice that co-overexpress human APP and presenilin 1 (PS1) harboring five familial AD (FAD) mutations under control of the neuron-specific Thy-1 promoter (Oakley et al, 2006; Ohno et al, 2006). 5XFAD mice start to develop visible amyloid deposition as early as 2 months of age consistent with their dramatically accelerated Aβ42 production because of a combination of the multiple FAD mutations, thus representing an early onset and aggressive amyloid mouse model (Oakley et al, 2006). At 4–6 months of age, 5XFAD mice start to show impairments in hippocampus-dependent learning and memory as tested by the Y-maze, Morris water maze, and contextual or trace fear conditioning paradigms (Kimura and Ohno, 2009; Oakley et al, 2006; Ohno et al, 2006). These behavioral deficits are observed concomitant with the onset of hippocampal CA1 synaptic dysfunctions such as reduced basal transmission and long-term potentiation (LTP: a synaptic plasticity model for learning and memory) (Kimura and Ohno, 2009). Furthermore, 5XFAD mice begin to exhibit elevations in expression levels of the β-secretase (β-site APP cleaving enzyme 1: BACE1), which is responsible for initiating the production of Aβ, at 6–9 months of age (Devi and Ohno, 2010b; Ohno et al, 2007; Zhang et al, 2009; Zhao et al, 2007), as observed in sporadic AD brains (Fukumoto et al, 2002; Li et al, 2004; Yang et al, 2003). To rigorously test the therapeutic potential of 7,8-DHF, we used the advanced stage of 5XFAD transgenic mice (12–15 months of age) that is more relevant to clinic AD cases manifesting robust amyloidosis, BACE1 elevations, BDNF-TrkB dysfunction, and profound memory impairments. The results provide novel and important insights into the use of 7,8-DHF or related small-molecule TrkB agonists as one of the promising and feasible approaches for BDNF-based AD therapy.
MATERIALS AND METHODS
Animals
We used 5XFAD transgenic mice (Tg6799 line) that co-express and co-inherit FAD mutant forms of human APP (the Swedish mutation: K670N, M671 L; the Florida mutation: I716V; the London mutation: V717I) and PS1 (M146 L; L286V) transgenes under transcriptional control of the neuron-specific mouse Thy-1 promoter (Oakley et al, 2006; Ohno et al, 2006). 5XFAD lines (B6/SJL genetic background) were maintained by crossing hemizygous transgenic mice with B6/SJL F1 breeders (Taconic, Hudson, NY). 5XFAD transgenic mice used were hemizygotes with respect to the transgene and non-transgenic wild-type littermate mice served as controls. Genotyping was performed by PCR analysis of tail DNA, as described previously (Oakley et al, 2006). Both male and female animals were used for the experiments as previous studies indicate that there is no sex difference in cerebral Aβ levels in 5XFAD mice except for the younger age (⩽3 months) (Oakley et al, 2006). All experiments were done blind with respect to the genotype of the mice. Procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Nathan Kline Institute Animal Care and Use Committee.
Drug Treatments
7,8-DHF (Sigma-Aldrich, St Louis, MO) was dissolved in phosphate-buffered saline (PBS) containing 17% dimethylsulfoxide (DMSO). 5XFAD transgenic and wild-type control mice at 12–15 months of age received repeated intraperitoneal injections of 5 mg/kg 7,8-DHF or 17% DMSO vehicle once daily for 10 consecutive days. Two hours after the last administration, the mice were subjected to behavioral testing. The dosage of 5 mg/kg 7,8-DHF and a 2-h interval between the last drug treatment and assays were chosen on the basis of previous in vivo studies (Andero et al, 2010; Andero et al, 2011; Jang et al, 2010; Liu et al, 2010), which demonstrated central TrkB activation, enhanced neurogenesis, and related behavioral changes in mice or rats treated with systemic 7,8-DHF administration.
Spontaneous Alternation Y-Maze Test
Spontaneous alternation performance was tested using a symmetrical Y-maze, as described previously (Kimura et al, 2010; Ohno et al, 2004). Each mouse was placed in the center of the Y-maze and was allowed to explore freely through the maze during an 8-min session. The sequence and total number of arms entered were recorded. Arm entry was considered to be complete when the hind paws of the mouse had been completely placed in the arm. Percentage alternation is the number of triads containing entries into all three arms divided by the maximum possible alternations (the total number of arms entered−2) × 100. As the reentry into the same arm was not counted for analysis, the chance performance level in this task was 50% in the choice between the arm mice visited more recently (non-alternation) and the other arm they visited less recently (alternation). After behavioral testing, some mice were killed for immunoblotting and ELISA experiments, and others were perfused for immunohistochemistry.
Immunoblot Analysis
Hemibrain or hippocampal samples were taken from the mice under deep isoflurane anesthesia and were snap-frozen for biochemical assays. For western blot analysis, each sample was homogenized in five volumes of modified RIPA buffer containing 150 mM NaCl, 50 mM Tris HCl (pH 8.0), 1 mM EDTA, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS, and protease/phosphatase inhibitor cocktail (Calbiochem, La Jolla, CA), and centrifuged at 10 000 g for 10 min to remove any insoluble material. Protein concentrations were determined by a BCA protein assay kit (Pierce, Rockford, IL), and 20–50 μg of protein was run on 4–12% NuPAGE gels (Invitrogen, Carlsbad, CA), and transferred to nitrocellulose membrane. After blocking, membranes were probed with anti-BDNF that is raised against the mature BDNF domain (130–247) (1 : 1000, sc-20981, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-TrkB that is raised against non-phosphopeptide derived from TrkB around the phosphorylation site of Tyrosine 515 (1 : 1000, ab51190, Abcam, Cambridge, MA, USA), an antibody against phosphorylated TrkB (Y705) (1 : 500, ab52191, Abcam), anti-BACE1 (1 : 1000, MAB5308, Millipore, Billerica, MA), an antibody that recognizes C-terminal epitope in APP (1 : 1000, C1/6.1, kindly provided by Dr Paul Mathews, Nathan Kline Institute) to detect full-length APP/C-terminal fragments, or anti-β-actin (1 : 15 000, AC-15, Sigma-Aldrich). They were then incubated with horseradish peroxidase-conjugated secondary IgG. Immunoblot signals were visualized by an ECL chemiluminescence substrate reagent kit (Pierce), and were quantified by densitometric scanning and image analysis using Quantity One software (Bio-Rad Laboratories, Hercules, CA).
Immunofluorescence Labeling
Mice were transcardially perfused with 0.1 M PBS (pH7.4), followed by 4% paraformaldehyde in PBS under deep isoflurane anesthesia. Brains were postfixed for 24 h in 4% paraformaldehyde in PBS at 4 °C, and transferred to PBS. The brain was sectioned coronally at 25 μm on a vibratome (VT1200, Leica Microsystems, Wetzlar, Germany), and successive sections were stored in PBS containing 0.01% sodium azide at 4°C. Two to three sections per mouse were taken at levels between –1.7 and –1.9 mm to bregma according to the mouse brain atlas of Franklin and Paxinos (2008), and incubated in blocking solution containing 10% purified goat serum and 0.05% Triton X-100 for 2 h. The sections were then incubated overnight at 4 °C in PBS containing a primary rabbit antibody against phosphorylated TrkB(Y705) (1 : 200, ab52191, Abcam), 10% goat serum, and 0.05% Triton X-100. Immunofluorescence labeling was performed by a 2-h reaction with Alexa Fluor 594-conjugated anti-rabbit IgG (1 : 500, Invitrogen) at room temperature. The sections were then washed three times in PBS and mounted with anti-fading medium. Control sections were processed with the omission of the primary antibody in the incubation buffer, and these controls yielded no specific labeling in brain sections. Immunostained sections were imaged with a confocal fluorescence microscope (LSM 510 Meta, Zeiss, Oberkochen, Germany) with a 40X objective.
ELISAs of Aβ40 and Aβ42
Sandwich Aβ ELISAs were performed as described previously (Devi and Ohno, 2010a; Devi and Ohno, 2010b; Kimura et al, 2010). Briefly, each hemibrain sample was extracted in 8X cold 5 M guanidine HCl plus 50 mM Tris HCl (pH 8.0) buffer, and centrifuged at 20 000 g for 1 h at 4°C to remove insoluble material. Final guanidine HCl concentrations were below 0.1 M. Protein concentrations were determined by a BCA kit (Pierce). To quantitate total levels of cerebral Aβ40 and Aβ42, supernatant fractions were analyzed by a well-established human Aβ40 and Aβ42 ELISA kits (KHB3481 and KHB3441, Invitrogen), respectively, according to the protocol of the manufacturer. Optical densities at 450 nm of each well were read on a VersaMax tunable microplate reader (Molecular Devices, Sunnyvale, CA), and sample Aβ40 and Aβ42 concentrations were determined by comparison with the respective standard curves. Aβ40 and Aβ42 concentration values were normalized to total brain protein concentrations and expressed as the percentage of vehicle controls.
In Vitro BACE1 Activity Assay
In vitro β-secretase activities were assessed by the SensiZyme BACE1 Activity Assay kit (CS1060, Sigma-Aldrich) according to the protocol of the manufacturer. Briefly, different concentrations of soluble recombinant BACE1 standard (0, 2.5, 5, and 10 ng/ml) were coincubated with 7,8-DHF or vehicle (0.25% DMSO). Using a proenzyme substrate that contains a BACE1-specific cleavage site, BACE1 activities were measured, in duplicate, as the absorbance at 405 nm on a VersaMax tunable microplate reader (Molecular Devices).
Statistical Analysis
Data were analyzed by a one-way or two-way ANOVA, and post-hoc Bonferroni comparisons were performed to determine the significance of differences between the groups when appropriate. Data were presented as mean±SEM, and the level of significance was set for p-value <0.05.
RESULTS
BDNF Declines Early During the Disease Progression in 5XFAD Mice
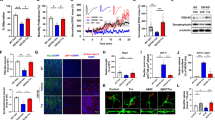
A growing body of evidence indicates that BDNF levels are decreased in brains of AD patients (Hu and Russek, 2008; Peng et al, 2005; Zuccato and Cattaneo, 2009) and APP transgenic mice (Francis et al, 2010; Peng et al, 2009). We first characterized changes in BDNF levels in the 5XFAD transgenic mouse model at different ages ranging from 3 to 18 months (Figure 1). Western blot analysis of hippocampal homogenate samples demonstrated dramatic reductions in baseline levels of mature BDNF (14 kDa) in 5XFAD mice (Figure 1a). A two-way ANOVA for mature BDNF levels revealed a significant main effect of genotype (F(1, 27)=56.34, p<0.05) in the absence of a main effect of age or a significant age × genotype interaction (Figure 1b). Post-hoc Bonferroni tests showed that mature BDNF levels were significantly reduced in 5XFAD mice at 3 months of age (p<0.05) as well as at 6 months and 15–18 months of age (p<0.05) compared with the respective age-matched wild-type controls. It has been reported that 3-month-old 5XFAD mice exhibit little or only faint amyloid pathology in brain (Devi et al, 2010; Oakley et al, 2006); therefore, our results are consistent with clinical observations that the reduction of mature BDNF starts to occur early in the course of AD (Peng et al, 2005).

Reductions of mature BDNF in 5XFAD mice. (a) Immunoblot analysis of hippocampal lysates from 5XFAD and age-matched wild-type control mice at different ages. (b) Immunoreactive bands for mature BDNF (14 kDa) were quantified and expressed as the percentage of wild-type control levels at 3 months of age (n=5–6 mice per group). Hippocampal mature BDNF levels are significantly reduced as early as 3 months of age in 5XFAD mice (*p<0.05 vs wild-type). All data are presented as mean±SEM.
Systemic 7,8-DHF Ameliorates Memory Deficits in 5XFAD Mice
To address the direct relationship between hippocampal BDNF deficiency and memory dysfunction, we examined whether systemic administration of the TrkB agonist 7,8-DHF could improve hippocampus-dependent memory decline in 5XFAD mice (Figure 2). 5XFAD mice at 12–15 months of age and their wild-type littermate controls received repeated injections of 7,8-DHF (5 mg/kg, i.p.) or vehicle (17% DMSO) once daily for 10 consecutive days, and were tested with the spontaneous alternation Y-maze paradigm that assesses spatial working memory function (Lalonde, 2002; Ohno et al, 2004). The dosage of 7,8-DHF was determined based on previous in vivo studies demonstrating that systemic administration of this compound activates central TrkB receptors and rescues memory deficits in BDNF-knockout mice (Choi et al, 2010; Jang et al, 2010). A two-way ANOVA revealed a significant genotype × drug interaction for the percent alternation performance (F(1, 28)=9.34, p<0.05) with a significant main effect of drug (F(1, 28)=9.51, p<0.05) (Figure 2a). The spontaneous alternation behavior relies on the inherent tendency of mice to enter a less recently visited arm compared with the other one; therefore, ∼67% alternation in wild-type controls reflects the mouse's ability to remember which arm they visited more recently during the successive arm entries in the Y-maze (spatial working memory). Levels of spontaneous alternation in vehicle-treated 5XFAD mice were reduced to ∼50% corresponding to the random performance level in this memory assay (p<0.05). Notably, post-hoc Bonferroni tests showed that the reduced spontaneous alternation was restored to wild-type control levels in 7,8-DHF-treated 5XFAD mice (p<0.05), whereas the alternation performance of wild-type mice was indistinguishable between 7,8-DHF- and vehicle-treated groups. Therefore, 7,8-DHF specifically improved the impaired spatial working memory in 5XFAD mice without affecting the normal working memory performance in wild-type mice (Figure 2a). Furthermore, neither significant genotype × drug interaction nor main effect of genotype or drug was found for exploratory activities as assessed by the number of arm entries during Y-maze testing (Figure 2b), indicating that the effect of 7,8-DHF in 5XFAD mice is memory-specific.

Effects of systemic 7,8-DHF on memory deficits in 5XFAD mice. 5XFAD and wild-type control mice at 12–15 months of age received repeated administration of 7,8-DHF (5 mg/kg, i.p.) or vehicle (17% DMSO) once daily for 10 consecutive days. Two hours after the last injection, the mice were tested for memory using the spontaneous alternation Y-maze task. (a) Spatial working memory, as assessed by the spontaneous alternation performance, is impaired (around 50% chance levels) in 5XFAD mice compared with wild-type controls (*p<0.05). Note that 7,8-DHF-treated 5XFAD mice are rescued completely back to wild-type levels of alternation performance (#p<0.05 vs vehicle-treated 5XFAD). n=5–12 mice per group. (b) Total number of arm entries reflecting exploratory activities of mice in the Y-maze does not differ between the groups, suggesting that the effect of 7,8-DHF is memory-specific. n=5–12 mice per group. All data are presented as mean±SEM.
Systemic 7,8-DHF Activates Hippocampal TrkB Receptors in 5XFAD Mice
To clarify whether hippocampal TrkB activation underlies the changes in memory function in 5XFAD mice, we conducted immunoblot analysis after behavioral testing (Figure 3a). We first measured levels of phosphorylated TrkB, an activated form of the TrkB receptor, in the hippocampus of mice treated with 7,8-DHF once daily for 10 days (Figure 3b). A two-way ANOVA for phospho-TrkB levels revealed a significant genotype × drug interaction (F(1, 24)=5.41, p<0.05) together with significant main effects of genotype (F(1, 24)=19.74, p<0.05) and drug (F(1, 24)=26.89, p<0.05). Post-hoc comparisons showed that levels of phosphorylated TrkB were significantly reduced in vehicle-treated 5XFAD mice compared with wild-type controls (∼9%) (p<0.05), whereas this reduction was rescued by 7,8-DHF treatments in 5XFAD mice (p<0.05). In fact, 7,8-DHF elevated the phosphorylation of TrkB in 5XFAD mice to levels that were significantly higher than those of wild-type vehicle controls (∼132%) (p<0.05). Meanwhile, systemic administration of 7,8-DHF induced robust activation of TrkB in wild-type mice as assessed by dramatic increases in phosphorylated TrkB levels compared with vehicle controls (∼423%) (p<0.05).

Effects of systemic 7,8-DHF on BDNF-TrkB signaling in 5XFAD mice. 5XFAD and wild-type control mice at 12–15 months of age received repeated administration of 7,8-DHF or vehicle (once daily for 10 consecutive days), and were killed 2 h after the last injection. (a) Immunoblot analysis of hippocampal lysates from wild-type and 5XFAD mice treated with 7,8-DHF or vehicle. (b–d) Immunoreactive bands for phosphorylated TrkB (b), total TrkB (c), and mature BDNF (d) were quantified and expressed as the percentage of vehicle-treated wild-type control levels (n=6–9 mice per group). Note that hippocampal phospho-TrkB levels are dramatically reduced in vehicle-treated 5XFAD mice (*p<0.05 vs wild-type, vehicle), and this reduction is completely rescued by 7,8-DHF treatments (#p<0.05 vs 5XFAD, vehicle). In fact, phospho-TrkB levels in 7,8-DHF-treated 5XFAD mice are significantly higher than those of wild-type vehicle controls (*p<0.05). Moreover, repeated 7,8-DHF treatments also rescues the downregulation of total TrkB protein levels in 5XFAD mice (#p<0.05); however, it shows a trend toward reducing TrkB receptors in wild-type controls (p=0.15). Meanwhile, 7,8-DHF treatments do not affect baseline levels of mature BDNF in the hippocampus of wild-type mice or BDNF reductions in 5XFAD mice. All data are presented as mean±SEM.
Interestingly, a two-way ANOVA for total TrkB protein levels also revealed a significant genotype × drug interaction (F(1, 24)=8.31, p<0.05) with a significant main effect of genotype (F(1, 24)=6.04, p<0.05) (Figure 3c). Post-hoc comparisons showed that levels of total TrkB in vehicle-injected 5XFAD were significantly reduced compared with wild-type controls (∼45%) (p<0.05), as is observed in AD brains (Ginsberg et al, 2010; Tapia-Arancibia et al, 2008). Notably, repeated administration of 7,8-DHF once daily for 10 days partially rescued the downregulation of hippocampal TrkB receptors found in 5XFAD mice (∼74%) (p<0.05). In contrast, 10-day treatments with 7,8-DHF showed a trend toward reducing total TrkB levels in wild-type mice (∼69%) (p=0.15), probably reflecting the adaptive downregulation of receptors in response to repeated exposures to the agonist. Meanwhile, a two-way ANOVA for mature BDNF levels showed only a main effect of genotype (F(1, 24)=70.06, p<0.05) in the absence of a main effect of drug or a significant genotype × drug interaction (Figure 3d). Post-hoc Bonferroni tests revealed that levels of mature BDNF were similarly reduced in the hippocampus of 5XFAD mice treated with vehicle (∼49%) (p<0.05) and 7,8-DHF (∼46%) (p<0.05), as compared with wild-type controls.
We further performed immunofluorescence staining of hippocampal sections to investigate TrkB phosphorylation provoked by systemic administration of 7,8-DHF in 5XFAD mice (Figure 4). Consistent with western blotting data, labeling for phosphorylated TrkB receptors was almost abolished in the hippocampal CA1 (Figure 4b) and CA3 (Figure 4e) regions of 5XFAD mice. Treatments with 7,8-DHF restored TrkB activation in both regions of 5XFAD mice (Figure 4c and f) almost completely back to wild-type control levels (Figures 4a and d). Collectively, the results clearly indicated that systemic administration of the TrkB agonist 7,8-DHF rescued deficient hippocampal BDNF-TrkB signaling through directly activating TrkB receptors in 5XFAD mice.

Immunofluorescence labeling of phosphorylated TrkB in the hippocampus of 5XFAD mice. 5XFAD mice at 12–15 months of age received repeated administration of 7,8-DHF or vehicle (once daily for 10 consecutive days), and were perfused for immunostaining 2 h after the last injection (n=3 mice per group). Shown are representative photomicrographs of hippocampal CA1 (a–c) and CA3 (d–f) regions. Note that phospho-TrkB staining is dramatically reduced in vehicle-treated 5XFAD mice (b, e) compared with vehicle-treated wild-type controls (a, d), whereas TrkB activation is restored back to wild-type levels in 7,8-DHF-treated 5CFAD mice (c, f). Scale bar=50 μm.
Systemic 7,8-DHF Reduces BACE1 and β-Amyloidogenesis in 5XFAD Mice
To investigate the possibility that repeated treatments with 7,8-DHF could improve memory through affecting APP processing in 5XFAD mice, we first conducted immunoblot analysis to assess levels of BACE1, full-length APP, and its β-cleaved fragments (Figure 5a). Previous studies from our laboratory and others have demonstrated that 5XFAD mice recapitulate significant elevations in BACE1 expression (Devi and Ohno, 2010b; Ohno et al, 2007; Zhang et al, 2009; Zhao et al, 2007) reminiscent of sporadic AD brains (Fukumoto et al, 2002; Li et al, 2004; Yang et al, 2003). A two-way ANOVA for BACE1 protein levels revealed significant main effects of genotype (F(1, 24)=66.38, p<0.05) and drug (F(1, 24)=30.69, p<0.05), but not a significant genotype × drug interaction (Figure 5b). Consistent with previous findings, post-hoc comparisons showed that BACE1 levels were dramatically increased in brains of vehicle-treated 5XFAD mice at 12–15 months of age compared with wild-type controls (∼166%) (p<0.05). Notably, repeated administration of 7,8-DHF once daily for 10 days significantly reduced BACE1 expression in 5XFAD brains (∼124%) (p<0.05). Repeated 7,8-DHF injections also significantly reduced BACE1 levels in wild-type control mice (∼41%) (p<0.05). Therefore, our results indicated that activation of the BDNF-TrkB signaling pathway not only suppresses BACE1 elevations in AD conditions but also downregulates baseline levels of BACE1 expression in normal brains.

Effects of systemic 7,8-DHF on APP processing and Aβ levels in 5XFAD mice. 5XFAD and wild-type control mice at 12–15 months of age received repeated administration of 7,8-DHF or vehicle (once daily for 10 consecutive days), and were killed 2 h after the last injection. (a) Immunoblot analysis of hemibrain lysates from wild-type and 5XFAD mice treated with 7,8-DHF or vehicle. (b, c) Immunoreactive bands for BACE1 (b) and C99 (c) were quantified and expressed as the percentage of vehicle-treated wild-type and 5XFAD levels, respectively (n=6–9 mice per group). Note that BACE1 levels are significantly elevated in vehicle-treated 5XFAD mice (*p<0.05 vs wild-type, vehicle), and this upregulation is suppressed by 7,8-DHF treatments (#p<0.05 vs 5XFAD, vehicle). Moreover, repeated 7,8-DHF treatment also significantly reduces baseline levels of BACE1 in wild-type mice (*p<0.05). Consistent with the BACE1 reduction, C99 levels are significantly lowered by 7,8-DHF treatments in 5XFAD mice (#p<0.05). (d, e) Levels of total Aβ40 (d) and Aβ42 (e) were quantified by sandwich ELISAs of guanidine extracts of hemibrain samples and expressed as the percentage of vehicle-treated 5XFAD levels (n=6–9 mice per group). Repeated 7,8-DHF treatment almost equivalently reduced Aβ40 and Aβ42 levels in 5XFAD mouse brains (#p<0.05 vs 5XFAD, vehicle). All data are presented as mean±SEM.
In accordance with reductions in BACE1, a one-way ANOVA showed that levels of the β-secretase-cleaved C-terminal fragment of APP (C99) in 7,8-DHF-treated 5XFAD mice were significantly lower than those of vehicle-treated 5XFAD controls (∼58%) (F(1, 13)=11.11, p<0.05) (Figure 5c). Meanwhile, transgene-derived overexpression of human full-length APP was not affected by repeated administration of 7,8-DHF in 5XFAD mice (Figure 5a). We further performed sandwich ELISAs to examine the effects of 7,8-DHF on cerebral Aβ levels (Figures 5d and e). Remarkably, repeated injections of 7,8-DHF once daily for 10 days significantly and almost equivalently lowered excessive levels of Aβ40 (∼47%) (F(1, 13)=14.17, p<0.05) and Aβ42 (∼44%) (F(1, 13)=8.06, p<0.05) without affecting the ratio between Aβ40 and Aβ42 (F(1, 13)=0.12, p=0.73) in 5XFAD mouse brains, which was consistent with the downregulation of BACE1 expression.
Effects of Acute 7,8-DHF Treatment on BACE1 Expression and Activity
As repeated administration of 7,8-DHF resulted in significant reductions in BACE1 expression, we examined whether an acute application of this compound may affect levels of BACE1 expression and/or enzymatic activity (Figure 6). A single injection of 5 mg/kg 7,8-DHF induced robust activation of TrkB receptors as measured by increases in phosphorylated TrkB (∼626%) (F(1, 6)=70.97, p<0.05) without affecting total TrkB levels in wild-type mice (Figure 6a). This observation supports the idea that reduced total TrkB in wild-type controls that received 10-day 7,8-DHF treatments (Figure 3c) may result from the compensatory downregulation of receptors in response to repeated exposures to the agonist. Notably, single 7,8-DHF administration was sufficient to reduce BACE1 expression levels in wild-type mouse brains (∼41%) (F(1, 6)=40.42, p<0.05) (Figure 6b), which was equivalent to the reductions following 10-day repeated injections (Figure 5b).

Effects of acute 7,8-DHF application on BACE1 expression and enzymatic activity. (a, b) Wild-type mice received single administration of 7,8-DHF (5 mg/kg, i.p.) or vehicle, and were killed 2 h later for the analysis. Immunoblot analysis shows that acute 7,8-DHF dramatically increases hippocampal phospho-TrkB levels (*p<0.05 vs vehicle) without affecting total TrkB (a). Note that a single injection of 7,8-DHF is sufficient to significantly reduce baseline levels of BACE1 in wild-type mouse brains (*p<0.05 vs vehicle) (b). n=4 mice per group. Data are presented as mean±SEM. (c) In vitro β-secretase activity assay was conducted by coincubating recombinant BACE1 with different concentrations of 7,8-DHF or vehicle. 7,8-DHF (1–10 μM) has no direct inhibitory effect on BACE1 activity.
To further test whether 7,8-DHF may directly inhibit BACE1 activity, we coincubated recombinant BACE1 with 7,8-DHF or vehicle (0.25% DMSO) in in vitro assays (Figure 6c). Enzyme activity was linear relative to the amount of recombinant BACE1 protein added. 7,8-DHF at concentrations of 1–10 μM had no effect on the catalytic activity of BACE1. Therefore, 7,8-DHF reduced BACE1 expression through activation of TrkB receptors, while this compound did not directly inhibit the enzymatic activity of BACE1.
DISCUSSION
We demonstrate that repeated systemic administration of the small-molecule TrkB receptor agonist 7,8-DHF rescues hippocampus-dependent memory deficits in the 5XFAD transgenic mouse model of AD. 5XFAD mice show impaired hippocampal BDNF-TrkB signaling, as measured by reductions in BDNF, TrkB receptors, and phosphorylated TrkB. Systemic 7,8-DHF can directly activate hippocampal TrkB, as evidenced by dramatic increases in TrkB phosphorylation without changes in endogenous BDNF levels, leading to complete reversal of deficient BDNF-TrkB signaling in 5XFAD mice. These findings provide a proof of concept that pharmacological activation of central TrkB receptors with non-peptide BDNF mimetic agents such as 7,8-DHF may represent an efficacious therapeutic approach to treat AD-related memory deficits. Aβ has been shown to decrease BDNF mRNA levels (Garzon and Fahnestock, 2007), impair BDNF retrograde trafficking (Poon et al, 2011), and downregulate BDNF-TrkB signaling pathways (Tong et al, 2004). It is also well documented that genetic reductions of BDNF or TrkB deteriorate basal synaptic transmission and/or LTP at the hippocampal Schaffer collateral-CA1 pathway and cause memory deficits in mice (Korte et al, 1995; Linnarsson et al, 1997; Minichiello, 2009; Minichiello et al, 1999; Pang and Lu, 2004; Patterson et al, 1996). Consistent with these results, we recently reported that partial reductions of Aβ induced by heterozygous BACE1 gene deletion (BACE1+/–) rescue synaptic and mnemonic dysfunctions in 5XFAD mice with the restoration of reduced hippocampal BDNF and TrkB phosphorylation to normal levels (Kimura et al, 2010). Taken collectively, our results indicate that BDNF-TrkB dysfunction is a crucial mechanism downstream to Aβ accumulation in 5XFAD mice, and that reversal of the deficient BDNF-TrkB signaling with 7,8-DHF may be sufficient to directly ameliorate memory impairments associated with AD.
When designing therapeutic strategies using BDNF or mimetic drugs for AD, one important consideration is the change of its receptor TrkB. TrkB levels are reported to decrease at the relatively early stage of AD (Ginsberg et al, 2010; Hu and Russek, 2008; Schindowski et al, 2008; Tapia-Arancibia et al, 2008; Zuccato and Cattaneo, 2009), which may be of concern to the potential limitation of BDNF therapy. Interestingly, we found that repeated treatments with the TrkB agonist 7,8-DHF (once daily for 10 days) partially but significantly rescued the decline in TrkB expression by itself in the hippocampus of 5XFAD mice. In contrast, the same repeated administration of 7,8-DHF significantly reduced total TrkB protein levels in the hippocampus of wild-type mice, probably reflecting the adaptive downregulation of BDNF receptors as a consequence of long-term activation with the agonist. Therefore, the upregulation of TrkB receptor expression occurs specifically in 5XFAD brains following repeated 7,8-DHF treatments. It seems reasonable to speculate that impaired BDNF signaling input may cause reductions in the TrkB receptor level during disease progression in 5XFAD mice, whereas pharmacological activation of TrkB receptors with 7,8-DHF may act to rescue the reduced expression of TrkB, allowing the greater activation of BDNF-TrkB signaling pathway for memory improvements in 5XFAD mice. Our results strongly support the idea that reductions in baseline levels of BDNF and TrkB associated with AD may be a critical underlying mechanism for deficits in learning and memory.
Our current findings with 7,8-DHF are in agreement with previous reports showing that central BDNF administration, gene delivery, or neural stem cell transplantation exerts neuroprotective effects by facilitating BDNF function and ameliorates learning and memory deficits in various animal models of AD including APP transgenic mice (Blurton-Jones et al, 2009; Nagahara et al, 2009). However, these therapeutic approaches need to overcome several problems to start the clinical translational research (Zuccato and Cattaneo, 2009); for example, the regulation of BDNF production and toxicity or mutagenesis potentially induced by the direct injection of viral vectors or neural stem cell lines into the brain parenchyma. In this regard, the use of 7,8-DHF or other systemically applicable small-molecule TrkB agonists may provide the more promising and feasible strategies to rescue memory impairments through restoring BDNF-TrkB dysfunction in AD and avoid the potential adverse effects associated with invasive methods of delivery or uncontrolled dosing.
It is also important to note that AD transgenic mice such as J20 and 3xTg-AD, which receive BDNF gene delivery or neuronal stem cell transplantation, show memory improvements in the absence of reductions in amyloid or tau-tangle pathology (Blurton-Jones et al, 2009; Nagahara et al, 2009). These results are in sharp contrast with our data that the repeated direct activation of TrkB with systemic 7,8-DHF produces significant reductions of BACE1 expression, and consequently of the β-secretase cleaved C-terminal fragment C99, in 5XFAD mice. Moreover, repeated administration of 7,8-DHF once daily for 10 days significantly reduced total Aβ40 and Aβ42 levels in brains of 5XFAD mice at the advanced stage suffering from severe amyloid plaque pathology. The discrepancy may arise from differences in transgenic AD mouse models used for the experiments and/or the extent to which individual treatments enhance BDNF-TrkB function. First, it is possible that pharmacological approaches with the potent agonist 7,8-DHF may activate BDNF-TrkB signaling more efficiently than gene delivery or stem cell strategies. Furthermore, previous studies from our laboratory and others have demonstrated that elevations in BACE1 have an important role in accelerating disease progression in 5XFAD mouse brains (Devi and Ohno, 2010b; O’Connor et al, 2008; Ohno et al, 2007; Zhang et al, 2009; Zhao et al, 2007) as well as in sporadic AD brains (Fukumoto et al, 2002; Li et al, 2004; Yang et al, 2003). In this study, TrkB phosphorylation was almost abolished (∼9%) while BACE1 expression was significantly elevated (∼166%) in 5XFAD mice compared with wild-type controls. Conceivably, robust TrkB activation, as evidenced by the increase in its phosphorylated active form that was higher than wild-type control levels (∼132%), enabled neurons to block BACE1 elevations in 7,8-DHF-treated 5XFAD mice. Multiple mechanisms are recently proposed to mediate the upregulation of BACE1 associated with AD. Those include the increased phosphorylation of the translation initiation factor eIF2α (Devi et al, 2010; Devi and Ohno, 2010b; O’Connor et al, 2008), caspase-3-dependent inactivation of GGA3 leading to decreased lysosomal degradation of BACE1 (Sarajarvi et al, 2009; Tesco et al, 2007), changes in microRNA expression profiles (Hebert et al, 2008; Wang et al, 2008), p25/cyclin-dependent kinase 5 pathways (Cruz et al, 2006; Wen et al, 2008), calpain activation (Liang et al, 2010), the receptor for advanced glycation end products (RAGE) (Cho et al, 2009; Guglielmotto et al, 2010), and oxidative stress or related signals such as nuclear factor-κB, c-Jun N-terminal kinase, and p38 MAPK (Chen et al, 2011; Chen et al, 2008; Coma et al, 2008; Xiong et al, 2007). Further study will be needed to determine the molecular pathways by which activation of BDNF-TrkB signaling may counteract the BACE1 elevation in 5XFAD mice. In any case, our findings suggest that long-term TrkB receptor activation with systemic 7,8-DHF can ameliorate memory deficits in 5XFAD mice, at least in part, through mediation of reductions in BACE1 expression and β-amyloidogenesis. Although the current study shows the positive effects of 7,8-DHF at the advanced phase of 5XFAD mice, it will be important to further evaluate this compound at different pathological stages and/or in other transgenic mouse models of AD.
It is hypothesized that Aβ accumulation induces BACE1 in plaque-surrounding neurons while the BACE1 elevation further accelerates Aβ production in 5XFAD mouse or AD brains at advanced stages of disease (Devi and Ohno, 2010b; Zhang et al, 2009; Zhao et al, 2007). Our results suggest that deficient BDNF-TrkB signaling is important for this positive feedback pathogenic mechanism in AD progression. However, it remains unclear whether BDNF-TrkB dysfunction may be involved in triggering AD at the incipient stage. To address this problem, we examined the effect of 7,8-DHF treatments on baseline levels of BACE1 expression in wild-type control mice. Remarkably, robust activation of TrkB receptors with repeated administration of 7,8-DHF induces significant reductions in BACE1 protein levels. Moreover, a single injection of 7,8-DHF is sufficient to similarly reduce BACE1 expression in brain. Meanwhile, in vitro assays revealed that 7,8-DHF does not have direct inhibitory effects on the enzymatic activity of BACE1. Together, the results indicate that the BDNF-TrkB pathway has a critical role in regulating baseline levels of BACE1 expression in normal animals, which is independent of interactions with human Aβ accumulation in disease conditions. It should be noted that levels of BDNF and TrkB decline in brains of patients with mild cognitive impairment or early AD (Ginsberg et al, 2010; Peng et al, 2005). Therefore, it seems likely that deficient BDNF-TrkB signaling pathways may not only be a mediator of memory impairments as a consequence of Aβ accumulation but also have an important role in initiating AD through the elevation of BACE1 expression. In this context, the use of TrkB receptor agonists may be beneficial for preventing or slowing down the development of AD at the incipient phase of disease.
In conclusion, our results strongly suggest that systemic administration of small-molecule TrkB receptor agonists such as 7,8-DHF may represent a promising and feasible therapeutic strategy to reverse deficient BDNF-TrkB signaling and memory impairments associated with AD. Although additional investigation is required, the present findings also provide preclinical evidence that activating TrkB receptors with BDNF mimetics downregulates BACE1 expression and β-amyloidogenesis, and thereby may serve as a disease-modifying approach in AD treatments.
References
Andero R, Daviu N, Escorihuela RM, Nadal R, Armario A (2010). 7,8-Dihydroxyflavone, a TrkB receptor agonist, blocks long-term spatial memory impairment caused by immobilization stress in rats. Hippocampus doi:10.1002/hipo.20906.
Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ (2011). Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry 168: 163–172.
Arancibia S, Silhol M, Mouliere F, Meffre J, Hollinger I, Maurice T et al (2008). Protective effect of BDNF against β-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol Dis 31: 316–326.
Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Muller FJ, Loring JF et al (2009). Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci USA 106: 13594–13599.
Chen CH, Zhou W, Liu S, Deng Y, Cai F, Tone M et al (2011). Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer's disease. Int J Neuropsychopharmacol 18: 1–14.
Chen L, Na R, Gu M, Richardson A, Ran Q (2008). Lipid peroxidation up-regulates BACE1 expression in vivo: a possible early event of amyloidogenesis in Alzheimer's disease. J Neurochem 107: 197–207.
Cho HJ, Son SM, Jin SM, Hong HS, Shin DH, Kim SJ et al (2009). RAGE regulates BACE1 and Aβ generation via NFAT1 activation in Alzheimer's disease animal model. FASEB J 23: 2639–2649.
Choi DC, Maguschak KA, Ye K, Jang SW, Myers KM, Ressler KJ (2010). Prelimbic cortical BDNF is required for memory of learned fear but not extinction or innate fear. Proc Natl Acad Sci USA 107: 2675–2680.
Coma M, Guix FX, Ill-Raga G, Uribesalgo I, Alameda F, Valverde MA et al (2008). Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiol Aging 29: 969–980.
Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT et al (2006). p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid β in vivo. J Neurosci 26: 10536–10541.
Devi L, Alldredand MJ, Ginsberg SD, Ohno M (2010). Sex- and brain region-specific acceleration of β-amyloidogenesis following behavioral stress in a mouse model of Alzheimer's disease. Mol Brain 3: 34.
Devi L, Ohno M (2010a). Genetic reductions of β-site amyloid precursor protein-cleaving enzyme 1 and amyloid-β ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer's disease model mice. Eur J Neurosci 31: 110–118.
Devi L, Ohno M (2010b). Phospho-eIF2α level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS One 5: e12974.
Francis BM, Kim J, Barakat ME, Fraenkl S, Yucel YH, Peng S et al (2010). Object recognition memory and BDNF expression are reduced in young TgCRND8 mice. Neurobiol Aging doi:10.1016/j.neurobiolaging.2010.1004.1003.
Franklin KBJ, Paxinos G (2008). The Mouse Brain in Stereotaxic Coordinates. Academic Press: New York.
Fukumoto H, Cheung BS, Hyman BT, Irizarry MC (2002). β-Secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol 59: 1381–1389.
Garzon DJ, Fahnestock M (2007). Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J Neurosci 27: 2628–2635.
Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y et al (2010). Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry 68: 885–893.
Guglielmotto M, Aragno M, Tamagno E, Vercellinatto I, Visentin S, Medana C et al (2010). AGEs/RAGE complex upregulates BACE1 via NF-κB pathway activation. Neurobiol Aging doi:10.1016/j.neurobiolaging.2010.1005.1026.
Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN et al (2008). Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/β-secretase expression. Proc Natl Acad Sci USA 105: 6415–6420.
Hu Y, Russek SJ (2008). BDNF and the diseased nervous system: a delicate balance between adaptive and pathological processes of gene regulation. J Neurochem 105: 1–17.
Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y et al (2010). A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci USA 107: 2687–2692.
Kimura R, Devi L, Ohno M (2010). Partial reduction of BACE1 improves synaptic plasticity, recent and remote memories in Alzheimer's disease transgenic mice. J Neurochem 113: 248–261.
Kimura R, Ohno M (2009). Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis 33: 229–235.
Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T (1995). Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA 92: 8856–8860.
Lalonde R (2002). The neurobiological basis of spontaneous alternation. Neurosci Biobehav Rev 26: 91–104.
Li R, Lindholm K, Yang LB, Yue X, Citron M, Yan R et al (2004). Amyloid β peptide load is correlated with increased β-secretase activity in sporadic Alzheimer's disease patients. Proc Natl Acad Sci USA 101: 3632–3637.
Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG (2010). Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Biol Chem 285: 27737–27744.
Linnarsson S, Bjorklund A, Ernfors P (1997). Learning deficit in BDNF mutant mice. Eur J Neurosci 9: 2581–2587.
Liu X, Chan CB, Jang SW, Pradoldej S, Huang J, He K et al (2010). A synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J Med Chem 53: 8274–8286.
Minichiello L (2009). TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 10: 850–860.
Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V et al (1999). Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24: 401–414.
Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM et al (2009). Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med 15: 331–337.
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J et al (2006). Intraneuronal β-amyloid aggregates neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26: 10129–10140.
O’Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL et al (2008). Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 60: 988–1009.
Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL et al (2006). Temporal memory deficits in Alzheimer's mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci 23: 251–260.
Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R et al (2007). BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis 26: 134–145.
Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M et al (2004). BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron 41: 27–33.
Pang PT, Lu B (2004). Regulation of late-phase LTP and long-term memory in normal and aging hippocampus: role of secreted proteins tPA and BDNF. Ageing Res Rev 3: 407–430.
Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER (1996). Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16: 1137–1145.
Peng S, Garzon DJ, Marchese M, Klein W, Ginsberg SD, Francis BM et al (2009). Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer's disease. J Neurosci 29: 9321–9329.
Peng S, Wuu J, Mufson EJ, Fahnestock M (2005). Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J Neurochem 93: 1412–1421.
Poon WW, Blurton-Jones M, Tu CH, Feinberg LM, Chabrier MA, Harris JW et al (2011). β-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol Aging 32: 821–833.
Sarajarvi T, Haapasalo A, Viswanathan J, Makinen P, Laitinen M, Soininen H et al (2009). Down-regulation of seladin-1 increases BACE1 levels and activity through enhanced GGA3 depletion during apoptosis. J Biol Chem 284: 34433–34443.
Schindowski K, Belarbi K, Buee L (2008). Neurotrophic factors in Alzheimer's disease: role of axonal transport. Genes Brain Behav 7 (Suppl 1): 43–56.
Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S (2008). New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev 59: 201–220.
Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M et al (2007). Depletion of GGA3 stabilizes BACE and enhances β-secretase activity. Neuron 54: 721–737.
Tong L, Balazs R, Thornton PL, Cotman CW (2004). β-Amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J Neurosci 24: 6799–6809.
Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q et al (2008). The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of β-site amyloid precursor protein-cleaving enzyme 1. J Neurosci 28: 1213–1223.
Wen Y, Yu WH, Maloney B, Bailey J, Ma J, Marie I et al (2008). Transcriptional regulation of β-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron 57: 680–690.
Xiong K, Cai H, Luo XG, Struble RG, Clough RW, Yan XX (2007). Mitochondrial respiratory inhibition and oxidative stress elevate β-secretase (BACE1) proteins and activity in vivo in the rat retina. Exp Brain Res 181: 435–446.
Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL et al (2003). Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 9: 3–4.
Zhang X-M, Cai Y, Xiong K, Cai H, Luo X-G, Feng J-C et al (2009). β-Secretase-1 elevation in transgenic mouse models of Alzheimer's disease is associated with synaptic/axonal pathology and amyloidogenesis: implications for neuritic plaque development. Eur J Neurosci 30: 2271–2283.
Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O’Connor T et al (2007). β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci 27: 3639–3649.
Zuccato C, Cattaneo E (2009). Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 5: 311–322.
Acknowledgements
This work was supported by grants from the Alzheimer's Association (IIRG-08-91231) and American Health Assistance Foundation (A2011311) to MO.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Devi, L., Ohno, M. 7,8-Dihydroxyflavone, a Small-Molecule TrkB Agonist, Reverses Memory Deficits and BACE1 Elevation in a Mouse Model of Alzheimer's Disease. Neuropsychopharmacol 37, 434–444 (2012). https://doi.org/10.1038/npp.2011.191
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.191
Keywords
This article is cited by
-
Brain-Derived Neurotrophic Factor: A Novel Dynamically Regulated Therapeutic Modulator in Neurological Disorders
Neurochemical Research (2023)
-
A TrkB agonist prodrug prevents bone loss via inhibiting asparagine endopeptidase and increasing osteoprotegerin
Nature Communications (2022)
-
Exercise Rehabilitation and/or Astragaloside Attenuate Amyloid-beta Pathology by Reversing BDNF/TrkB Signaling Deficits and Mitochondrial Dysfunction
Molecular Neurobiology (2022)
-
The tyrosine kinase inhibitor LPM4870108 impairs learning and memory and induces transcriptomic and gene‑specific DNA methylation changes in rats
Archives of Toxicology (2022)
-
Investigation of the Molecular Role of Brain-Derived Neurotrophic Factor in Alzheimer’s Disease
Journal of Molecular Neuroscience (2022)
