
Abstract
By the acute stage of HIV-1 infection, the immune system already faces daunting challenges. Research on mucosal barriers and the events immediately after heterosexual transmission that precede this acute stage could facilitate the development of effective microbicides and vaccines.
Similar content being viewed by others

Main
The AIDS epidemic has exacted a terrible toll in terms of loss of life and decreased quality of life worldwide, especially in Africa, where 70% of deaths from HIV-1 infection have occurred1. AIDS increasingly has a woman's face; almost 60% of individuals infected with HIV-1 are women2. There is therefore a great urgency to develop vaccines, microbicides and other preventive strategies to blunt the progress of the epidemic and, in particular, to stop the transmission of HIV-1 to women.
This review focuses on studies of this predominant route of heterosexual transmission, as well as the acute stage of infection by HIV-1 and the closely related simian immunodeficiency virus (SIV). Because transmission and the earliest stages of infection cannot be investigated systematically in humans, the in vivo studies that we discuss are conducted in rhesus macaques inoculated intravaginally and atraumatically with SIV. Although far from perfect, the many parallels in anatomy, physiology, immunology and nature and course of infection make this model, in our view, the most relevant for in vivo study3,4.
The main lesson to be learned from the HIV-1 and SIV studies is how quickly the virus establishes persistent infection in a lymphatic tissue reservoir and compromises host defenses. Because of this, we conclude that microbicides and vaccines that elicit durable mucosal immunity, thereby targeting the earliest stages of infection, have the best prospects for preventing or containing infection.
Acute HIV-1 infection: the die is cast
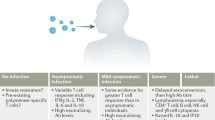
By the acute stage of HIV-1 infection, the immune system is already facing un uphill struggle. In the 2- to 3-week incubation period that follows sexual mucosal transmission (or transmission by oral or parenteral routes), the virus becomes well established in a lymphatic tissue reservoir5. This reservoir6,7,8,9,10,11,12 is the principal site of virus production, storage, persistence (Fig. 1) and pathology13 (CD4+ T-cell depletion and destruction of follicles and the lymphatic tissue architecture). By the time the infection becomes symptomatic in the clinical illness associated with seroconversion14,15—most commonly fever, skin rash, oral ulcers and lymphadenopathy16,17,18,19—the lymphatic tissues teem with productively, chronically and latently infected cells, and with virions (Fig. 1).

Katie Ris
In HIV-1 (and SIV) infections, the lymphatic tissues are the reservoir where virus is produced and stored in immune complexes bound by FDCs, and where virus persists in latently infected resting CD4+ T cells. Viral replication generates antigenic escape mutants and depletes CD4+ T cells (including HIV-1-specific CD4+ T cells) either directly or indirectly. FDCs can also reactivate infectious virus from the large FDC stores. Collectively, these obstacles to the elimination or control of infection by host defenses are already established in the acute stages of infection.
From this acute stage until the late stages of infection, billions of virions are produced daily, mainly by activated CD4+ T cells20. About 10–100 million of the infected activated CD4+ T cells die each day21 in a dynamic process22,23 in which each infected cell in the acute stage of infection generates about 20 infected progeny during its lifetime24. Once acute infection is established, a cellular immune response to HIV-1 is generated that partially controls viral replication25. It has been estimated, however, that a vaccine of at least 95% efficacy would be required to extinguish productive infection of this magnitude in the acute stage24.
In addition to the general loss of CD4+ T cells, the virus preferentially infects and eliminates HIV-1-specific CD4+ T cells26, which further compromises the virus-specific immune response. In addition, the error-prone viral replication process generates mutants that can escape neutralizing antibodies27,28 and virus-specific cytotoxic T lymphocytes (CTLs) in both HIV-1 and SIV infections29,30,31,32, further contributing to the continuing difficulties that the immune system encounters in containing infection.
The lymphatic tissues also contain a population of infected resting CD4+ T cells20, variously estimated at 1–100 million11,20,33, and a large pool of 5–50 billion virions in immune complexes bound to follicular dendritic cells (FDCs)9,10,11,12,34 in the acute and subsequent stages of infection. The infected resting CD4+ T cells either produce small quantities of virus or are latently infected. Infectious virus can be produced by the activation of latently infected cells and by the reactivation of virus in immune complexes by FDCs. Thus, there are two long-lived reservoirs and sources of virus that persist despite host defenses and current antiretroviral therapy (ART) and that can refuel infection35,36. Thus, by the time there is any indication of infection, the immune system, vaccines or ART are already faced with huge obstacles to the eradication or full control of infection.
The early stages of HIV-1 transmission
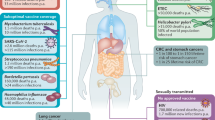
The litany of difficulties that host defenses confront in acute infection points to the importance of analyzing even earlier events in infection to discover where the virus may be more vulnerable to sterilizing immunity or to other means of preventing infection. Below, we discuss some of the issues relevant to the early stages of pathogenesis, from infection at the portal of entry to dissemination and establishment of a persistent infection in the lymphatic tissues (Fig. 2). Our discussion focuses on the transmission of cell-free virus because so much more is known about the transmission in vivo of cell-free as opposed to cell-associated virus, but there are probably inherent differences in how cell-associated viruses are transmitted.

Katie Ris
Shown are the major stages and events in transmission relevant to development of microbicides, vaccines and other preventive measures. R5 viruses from a viral quasispecies of R5 and X4 viruses are selectively transmitted. After crossing the cervicovaginal mucosal barrier, DCs, CD4+ T cells and macrophages in the underlying submucosa are infected. Infection is subsequently propagated and disseminated, thereby establishing the lymphatic tissue reservoir that spreads infection to other organs and peripheral tissues. Innate and adaptive host defenses (left column) are directed at the different stages to prevent transmission and contain infection.
Viral dose and transmission. The probability of transmission for each encounter with HIV-1 is quite small (about 0.001) and is related, in part, to dose37. There is a direct relationship between viral load in peripheral blood and transmission, with a cutoff of around 1,500 copies of viral RNA per ml of blood, below which there is no evidence of transmission38. The extent to which the concentration of virus in semen mirrors that in peripheral blood is unknown, but this dose dependency in transmission suggests that ART and vaccines that lower viral load might have a significant impact in reducing transmission at the population level.
The relationship between dose and transmission highlights one of the principal imperfections of the nonhuman primate models: the dose of SIV required to infect macaques consistently by the intravaginal route, about 105 TCID50 (50% tissue culture infectious doses)3,4, is probably far greater than that required to infect humans and may skew the appraisal of vaccine (and microbicide) candidates in the macaque model towards an underestimation of their potential efficacy.
R5 and X4 viruses. Transmission of HIV-1 to CD4+ T cells is mediated by interactions between the viral envelope glycoproteins gp120 and gp41, the T-cell receptor CD4 and the chemokine coreceptors CCR5 (ref. 39) and CXCR4 (ref. 40). During the course of HIV-1 infection, the error-prone viral polymerase generates genetically diverse quasispecies, including the strains designated R5 and X4, which use the CCR5 and CXCR4 coreceptors, respectively, to infect cells41.
Mainly R5 strains are transmitted, for reasons discussed below, whereas X4 strains emerge later and are associated with a rapid progression to AIDS42. A deletion of 32 base pairs in CCR5 prevents HIV-1 infection43,44,45, which indicates that there is strong selection for the R5 strain during transmission and early infection.
Crossing the mucosal barrier
Inflammation, menstrual cycle and hormones. The idealized intact vaginal epithelium contains stratified squamous cells, several layers thick, and an intact single layer of columnar epithelium in the endocervix that may be more easily traversed by HIV-1 and SIV (Fig. 2). In reality, the vaginal epithelial barrier changes markedly during the menstrual cycle and in response to exogenously administered hormones such as progesterone46. Pre-existing inflammatory conditions such as sexually transmitted diseases, bacterial vaginosis and some microbicides (such as nonoxynol-9) also cause microulcerations and breaks in the epithelium.
Thinning and breaches in the mucosal barrier, created in the above ways, could quickly expose susceptible cells in the submucosa to virus. This would increase the likelihood of establishing and rapidly disseminating infection, and would decrease the opportunities for a microbicide to inactivate the virus or for a vaccine-induced recall response to contain the infection at the portal of entry. In SIV infection, this could be the basis for the reported resistance to inactivation of the inoculated virus within 1 h and rapid dissemination beyond the portal of entry within 24 h (ref. 47), and may also be responsible for enhanced intravaginal transmission in monkeys pretreated with progesterone. In HIV-1 infection, thinning and breaches may underlie the increased transmission associated with sexually transmitted diseases, bacterial vaginosis and nonoxynol-9 use48,49,50,51, as well as the association between cervical ectopy and increased HIV-1 transmission52.
It may be easier for viruses to traverse the single layer of epithelium that lines the endocervix than the multilayer squamous epithelium of the ectocervix and vaginal mucosa. This would account for both the association between cervical ectopy and increased HIV-1 transmission52, and the higher concentration of SIV-infected cells found in the cervical submucosa after intravaginal inoculation20. The fact that the virus crosses the mucosal barrier so rapidly and gains access to dendritic cells (DCs) and CD4+ T cells to facilitate virus production and dissemination limits opportunities for a vaccine-induced anamnestic response to HIV-1 that could prevent or fully control infection.
Mechanisms for crossing an intact mucosal barrier. HIV-1 could traverse the intact stratified squamous epithelium of the vaginal mucosa and the simple columnar epithelium of the cervix by the modes of transmission and dissemination shown in Fig. 3. Transcytosis by a vesicular pathway is one mechanism by which HIV-1 could cross an intact barrier to infect susceptible host cells in the underlying tissues53, but it is also a stage at which the virus is vulnerable to mucosal antibody responses because secretory antibodies neutralize the infectivity of transcytosing virus54,55. In addition, SIV bound by neutralizing antibodies specific for the viral envelope is also significantly less effective than is unbound virus at infecting macaques through the vaginal route56.

Katie Ris
Viruses cross the barrier by capture or infection of DCs (1), transcytosis (2) or infection of intraepithelial lymphocytes (3). After R5 virus is ferried across the barrier or produced by DCs or intraepithelial lymphocytes, it infects resting and activated CD4+ T cells, DCs, DC and T-cell conjugates, macrophages, and macrophage and T-cell conjugates. There is also enhanced transmission to T cells by viruses captured by DCs through CLRs. Virus produced by T cells, DCs, macrophages and infected cells spread the infection to the draining lymph nodes. Infection may be initially confined to the portal of entry with subsequent dissemination, or virions and infected cells may rapidly spread infection to the draining lymph nodes and beyond.
Epithelial cells selectively capture R5 HIV-1 and then transfer infection to CCR5-expressing target cells underneath the epithelia, which could account for the preferential transmission of HIV-1 strain R5 (ref. 57). Dendritic cells positioned in the stratified squamous epithelium of the vaginal mucosa (with processes that extend to the lumenal surface to sample contents and to trap pathogens) and in the underlying tissues of the vagina and cervix are also prime targets for HIV-1 (Fig. 3). Dendritic cells express CD4, CCR5, DC-SIGN58 and other C-type lectin receptors (CLRs) that facilitate capture and infection by HIV-1 and SIV59,60. Different subsets of DCs express unique members of the CLR family, but the virus can use specific CLRs on each subset to ensure capture and dissemination. For example, DCs in vaginal epithelium express langerin (also known as CD207) but lack DC-SIGN (CD209) and mannose receptor (CD206), whereas DCs in the lamina propria express DC-SIGN and mannose receptor but not langerin, and yet both subsets bind virus in a CLR-dependent manner61.
Evidence indicates that in vivo, both intraepithelial and submucosal DCs and CD4+ T lymphocytes are the predominant cell populations first targeted by SIV and HIV-1. After intravaginal inoculation of SIV, viral DNA and RNA are detected within 24–72 h of exposure in DCs47,62, intraepithelial lymphocytes and CD4+ T cells (mainly resting, but also activated) in the endocervical submucosa20. In female genital organ cultures ex vivo, the first HIV-1-infected cells that could be detected were intraepithelial memory CD4+ T cells63,64.
Local propagation and dissemination
After crossing the epithelial barrier, HIV-1 and SIV can infect CCR5-expressing DCs, macrophages and T cells in the underlying mucosal tissues (Figs. 2 and 3) to initiate infection. Dendritic cells capture HIV-1 through CLRs61 and are productively infected by a CCR5-dependent mechanism65,66. Captured virus can be internalized (without productive infection) and then rapidly transmitted to nearby CD4+ T cells within an hour, in the form of an 'infectious synapse'67 (S.G. Turville et al., unpublished data). Productive infection in CD4+ T cells is facilitated in these DC–T cell conjugates68,69 and can spread infection to more CD4+ T cells, most efficiently to the memory subset59, which is the chief producer of virus in vivo20. In carrying out their normal function of conveying pathogens and antigens to the draining lymphatic tissues to induce an immune response, virus-carrying DCs can also disseminate infection to large numbers of CD4+ T cells to amplify and further disseminate the infection.
Transmitted strains of HIV-1 and SIV were originally referred to as macrophage- or M-tropic because the viruses replicated in vitro in monocyte and macrophage cultures but not in T-cell lines70. The tropism was subsequently shown to be a consequence of selection for R5 viral strains. In fact, there is no correlation in vivo between M-tropism and intravaginal transmission of SIV4. But infection of macrophages and DCs may be important in transmission by recruiting and enhancing virus production in T cells in ways that circumvent the activation of antiviral T-cell responses. Viral replication and viral gene products (including Nef, Tat and Vpx)71,72,73,74,75,76 induce macrophages and DCs to forge contacts with T cells and to drive the productive infection of resting and activated CD4+ T cells. This may partly explain the predominance of infection in resting CD4+ T cells during early SIV infection after intravaginal inoculation20 and in genital organ cultures in vitro63,64.
HIV-1 transmission in vivo
Understanding which coreceptors and other molecules are involved in virus-host cell interactions and how these interactions occur helps to identify the kinds of cells that HIV-1 (or SIV) can infect. But it does not tell us which types of cells, and in what proportions, will actually be infected either at transmission or in the acute and later stages of infection in an affected individual. These factors may be determined by substrate availability77.
The types and proportions of cells infected in vivo may be determined by substrate availability and spatial proximity. In predator-prey models78, the likelihood of infecting a cell is determined by the density of a particular type of cell and its proximity to a virus-producing cell. Such a model can account for both the predominance of infection of CD4+ T cells at transmission and in acute and later stages of SIV and HIV-1 infection12,20, and the predominance of productive infection in ostensibly resting CD4+ T cells in the first two weeks of SIV infection, during which more than 80% of the cells containing SIV RNA are resting CD4+ T cells20.
In the acute stage of HIV-1 infection, roughly equal proportions of the predominant population of CD4+ T cells positive for HIV-1 RNA were typed as resting or activated20. At later stages, most of the infected CD4+ T cells were activated, consistent with the greater availability of activated CD4+ T cells, owing to chronic immune activation, to be infected. In addition, the clustering of SIV-infected cells one and two cell diameters away from a 'parent' infected cell79 and the clustering of SIV genotypes80 are consistent with the spatial proximity tenet of such a predator-prey model.
Virus production by resting and activated CD4 + T cells
Resting CD4+ T cells infected with SIV or HIV-1 have about five times fewer copies of viral RNA than do infected activated CD4+ T cells20, consistent with the low amount of virus production in this type of cell. But because CD4+ T cells predominate at transmission and during the first days of SIV infection, they could have key roles in spreading infection and in maintaining an unbroken chain of transmission from one infected cell to the next. The function of the infected activated cell may be to act as an amplifier, propagating infection to more cells at greater distances.
Staged versus rapid spread beyond the portal of entry
The answers to questions of how and when infection is disseminated from the portal of entry have important implications for vaccine development. On the one hand, if the virus replicates locally for several days at the portal of entry in small numbers of cells with low levels of virus production, a vaccine-induced memory response will have the best chance of containing and perhaps eliminating infection. If, on the other hand, the virus quickly reaches large numbers of CD4+ T cells in the draining lymph nodes and large amounts of virus are produced to disseminate infection systemically throughout the lymphatic tissues, the immune system will face an uphill battle, even with a rapid recall response. In addition, immune defenses will be further weakened by the loss of CD4+ T cells to infection. In SIV infection, these losses are very substantial, particularly in gut-associated lymphoid tissues81 where the largest populations of lymphocytes are located, but also at the portal of entry82.
Evidence from the SIV macaque model supports both schemes. There are reports that SIV replication is restricted to small foci in the cervical submucosa in the first week after intravaginal inoculation20, and that local amplification and 'stepwise' dissemination occurs after intrarectal transmission of SIV71. By contrast, within 24 h of intravaginal exposure, SIV-infected cells have been detected in the draining lymph nodes of SIV-infected rhesus macaques47, and within 48 h in pig-tailed macaques inoculated intravaginally with SIV/HIV-1 (ref. 83). These two observations might reflect, for example, a pre-existing inflammation that facilitates the rapid spread of virus. Whatever the mechanisms, they need to be understood if we are to devise countering strategies against rapid viral spread.
The race between virus and immune defenses: Too little, too late
Figure 2 characterizes the fight between virus and host as a struggle in part for dominance in what we call the 'in vivo effector-to-target ratio' (the ratio of virus-specific CD8+ T cells to productively infected cells). The host defenses will win if virus-specific CTLs quickly outnumber infected target cells, particularly at the portal of entry, whereas the virus will win if this is not the case. In both published and unpublished studies of the natural history of sexual mucosal transmission, it seems likely that the virus usually wins because the host is unable to mount a cellular immune response of sufficient magnitude, breadth and rapidity to contain infection.
After intrarectal or intravaginal inoculation of SIV, significant numbers of SIV-specific CD8+ T cells are not detected until the second to third week of infection, and the CD8 response is largely focused on immunodominant epitopes in Gag and Tat84. By this stage, there are large numbers of productively infected cells throughout the lymphatic tissues and an effector-to-target ratio of <1 at most sites (calculated as the ratio of Gag and Tat tetramer–positive CD8+ T cells to SIV RNA–positive cells; M. Reynolds et al., unpublished data). The relatively unchecked replication also quickly spawns the above-mentioned CTL escape mutants29,30,31,32, further compounding the difficulties of controlling infection.
Microbicide and vaccine success stories
These natural history studies paint a rather depressing picture of how quickly conditions become unfavorable for host defenses to prevent or clear infection, but there are success stories of prevention and reports of resistance to infection that provide proof of principal that sexual mucosal transmission can be stopped.
First, although antibodies are generated too late in the natural history of acute infection to contribute to host defenses, they might be more effective if induced by a vaccine or supplied locally in sufficient concentration to block entry and spread. Antibodies can block infection, as substantiated by the report that antiviral antibodies in a microbicide prevent infection by SIV56. Second, there are reports that vaccines can prevent transmission of intravaginally and intrarectally inoculated SIV85,86,87,88,89,90. Finally, some individuals who have been repeatedly exposed to HIV-1 remain seronegative and apparently uninfected, although the virus may be present at extraordinarily low levels; this resistance is correlated with a virus-specific cellular immune response91,92. Although the resistance may not be sustained—for example, exposed and uninfected women who leave and then return to commercial sex work can become infected—it suggests that a vaccine-induced durable local mucosal immune response may prevent infection.
Future research
When we look at the overview of transmission and acute infection (Figs. 1 and 2) and consider the great difficulties that host defenses encounter once the lymphatic tissue reservoir is established, we are persuaded that the primary focus of future studies for preventive strategies needs to be at the beginning of transmission—that is, at the mucosal barrier and the initial interaction of virus with host cells and host defenses. As far as the mucosal barrier is concerned, we need to understand the causes of pre-existing inflammation and how to treat it, and to develop effective microbicides that inactivate virus without destroying the integrity of the barrier. At and beyond the mucosal barrier, we need to examine the role of innate defenses in preventing or controlling infection in vivo.
Greater knowledge of these first-line defenses might lead to new measures to inactivate or inhibit replication of the virus before or immediately after it crosses the barrier. For effective vaccine development, we need to know how to elicit a durable cellular and humoral mucosal immune response comprising persistent antibodies in cervical vaginal fluids and virus-specific memory CD8+ T cells, both of which are needed to generate a rapid recall response at the portal of entry. Finally, we need a widely available nonhuman primate model of SIV infection that more closely resembles HIV-1transmission by repeated low-dose exposure, to look for correlates of protection and critical vaccine components.
In this review, we have focused on the early events of mucosal spread in women, but future studies will clearly also need to address parenteral, rectal, oral and mother-to-child transmission. Collectively, such studies will advance in important ways the urgent quest to develop effective microbicides and vaccines to reduce the spread of HIV-1 around the world.
References
World Health Organization (WHO). AIDS epidemic update: December 2002 (WHO, Geneva, 2002).
Annan, K.A. In Africa, AIDS has a woman's face. New York Times 29 December (2002), p.9.
Miller, C.J. et al. Genital mucosal transmission of simian immunodeficiency virus: animal model for heterosexual transmission of human immunodeficiency virus. J. Virol. 63, 4277–4284 (1989).
Miller, C.J. et al. In vivo replication capacity rather than in vitro macrophage tropism predicts efficiency of vaginal transmission of simian immunodeficiency virus or simian/human immunodeficiency virus in rhesus macaques. J. Virol. 72, 3248–3258 (1998).
Haase, A.T. Population biology of HIV-1 infection: viral and CD4+ T cell demographics and dynamics in lymphatic tissues. Annu. Rev. Immunol. 17, 625–656 (1999).
Tenner-Racz, K. et al. HTLV-III/LAV viral antigens in lymph nodes of homosexual men with persistent generalized lymphadenopathy and AIDS. Am. J. Pathol. 123, 9–15 (1986).
Biberfeld, P. et al. HTLV-III expression in infected lymph nodes and relevance to pathogenesis of lymphadenopathy. Am. J. Pathol. 125, 436–442 (1986).
Pantaleo, G. et al. Lymphoid organs function as major reservoirs for human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 88, 9838–9842 (1991).
Fox, C.H. et al. Lymphoid germinal centers are reservoirs of human immunodeficiency virus type 1 RNA. J. Infect. Dis. 164, 1051–1057 (1993).
Pantaleo, G. et al. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature 362, 355–358 (1993).
Embretson, J. et al. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature 362, 359–362 (1993).
Schacker, T. et al. Rapid accumulation of HIV in lymphatic tissue reservoirs during acute and early HIV infection: implications for timing of antiretroviral therapy. J. Infect. Dis. 181, 354–357 (2000).
Racz, P. et al. The spectrum of morphologic changes in lymph nodes from patients with AIDS or AIDS-related complex. Prog. Allergy 37, 81–181 (1985).
Cooper, D.A. et al. Acute AIDS retrovirus infection. Definition of a clinical illness associated with seroconversion. Lancet 1, 537–540 (1985).
Ho, D.D. et al. Primary human T-lymphotropic virus type III infection. Ann. Intern. Med. 103, 880–883 (1985).
Schacker, T.W., Hughes, J.P., Shea, T., Coombs, R.W. & Corey, L. Biological and virologic characteristics of primary HIV infection. Ann. Intern. Med. 128, 613–620 (1998).
Kahn, J.O. & Walker, B.D. Acute human immunodeficiency virus type 1 infection. N. Engl. J. Med. 339, 33–39 (1998).
Kinloch-de Loes, S. et al. Symptomatic primary infection due to human immunodeficiency virus type 1: review of 31 cases. Clin. Infect. Dis. 17, 59–65 (1993).
Lavreys, L. et al. Primary human immunodeficiency virus type 1 infection: clinical manifestations among women in Monbasa, Kenya. Clin. Infect. Dis. 30, 486–490 (2000).
Zhang, Z.-Q. et al. Sexual transmission and propagation of simian and human immunodeficiency viruses in two distinguishable populations of CD4+ T cells. Science 286, 1353–1357 (1999).
Cavert, W. et al. Kinetics of response in lymphoid tissues to antiretroviral therapy of HIV-1 infection. Science 276, 960–964 (1997).
Wei, X. et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373, 117–122 (1995).
Ho, D.D. et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123–126 (1995).
Little, S.J., McLean, A.R., Spina, C.A., Richman, D.D. & Havlir, D.V. Viral dynamics of acute HIV-1 infection. J. Exp. Med. 190, 841–850 (1999).
Koup, R.A. et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68, 4650–4655 (1994).
Douek, D.C. et al. HIV preferentially infects HIV-specific CD4+ T cells. Nature 417, 95–98 (2002).
Lewis, J. et al. Development of a neutralizing antibody response during acute primary human immunodeficiency virus type 1 infection and the emergence of antigenic variants. J. Virol. 72, 8943–8951 (1998).
Richman, D.D., Wrin, T., Little, S.J. & Petropoulos, C.J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 100, 4144–4149 (2003).
Goulder, P.J.R. et al. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3, 212–217 (1997).
Borrow, P. et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3, 205–211 (1997).
Price, D.A. et al. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 94, 1890–1895 (1997).
Allen, T.M. et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nat. Med. 407, 386–390 (2000).
Chun, T.-W. et al. Quantitation of latent tissue reservoirs and total body load in HIV-1 infection. Nature 387, 183–188 (1997).
Haase, A.T. et al. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274, 985–989 (1996).
Persaud, D., Zhou, Y., Siliciano, J.M. & Siliciano, R.F. Latency in human immunodeficiency virus type 1 infection: no easy answers. J. Virol. 77, 1659–1665 (2003).
Heath, S.L., Tew, J.G., Szakal, A.K. & Burton, G.F. Follicular dendritic cells and human immunodeficiency virus infectivity. Nature 377, 740–744 (1995).
Gray, R.H. et al. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai, Uganda. Lancet 357, 1149–1153 (2001).
Quinn, T.C. et al. Viral load and heterosexual transmission of human immunodeficiency virus type 1. N. Engl. J. Med. 342, 921–929 (2000).
Deng, H.K. et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381, 661–666 (1996).
Alkhatib, G. et al. CC CKR5: A RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272, 1955–1958 (1996).
Berger, E.A. et al. A new classification for HIV-1. Nature 391, 240 (1998).
Schuitemaker, H. et al. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus populations. J. Virol. 66, 1354–1360 (1992).
Paxton, W.A. et al. Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposures. Nat. Med. 2, 412–417 (1996).
Liu, R. et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 86, 367–377 (1996).
Dean, M. et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science 273, 1856–1861 (1996).
Marx, P.A. et al. Progesterone implants enhance SIV vaginal transmission and early virus load. Nat. Med. 2, 1084–1089 (1996).
Hu, J., Gardner, M.B. & Miller, C.J. Simian immunodeficiency virus rapidly penetrates the cervicovaginal mucosa after intravaginal inoculation and infects intraepithelial dendritic cells. J. Virol. 74, 6087–6095 (2000).
Martin, H.L. et al. Vaginal lactobacilli, microbial flora, and risk of human immunodeficiency virus type 1 and sexually transmitted disease acquisition. J. Infect. Dis. 180, 1863–1868 (1999).
Sweankambo, N. et al. HIV-1 infection associated with abnormal vaginal flora morphology and bacterial vaginosis. Lancet 350, 546–550 (1997).
Cohn, M.A. et al. Chronic inflammation with increased human immunodeficiency virus (HIV) RNA expression in the vaginal epithelium of HIV-infected Thai women. J. Infect. Dis. 184, 410–417 (2001).
Van Damme, L. et al. Effectiveness of COL-1492, a nonoxynol-9 vaginal gel, on HIV-1 transmission in female sex workers: a randomized controlled trial. Lancet 360, 971–977 (2002).
Moss, G.B. et al. Association of cervical ectopy with heterosexual transmission of human immunodeficiency virus: results of a study of couples in Nairobi, Kenya. J. Infect. Dis. 164, 588–591 (1991).
Bomsel, M. Transcytosis of infectious human immunodeficiency virus across a tight human epithelial cell line barrier. Nat. Med. 1, 42–47 (1997).
Bomsel, M. et al. Intracellular neutralization of HIV transcytosis across tight epithelial barriers by anti-HIV envelope protein dIgA or IgM. Immunity 9, 277–287 (1998).
Belec, L. et al. Cervicovaginal secretory antibodies to human immunodeficiency virus type 1 (HIV-1) that block viral transcytosis through tight epithelial barriers in highly exposed HIV-1-seronegative African women. J. Infect. Dis. 184, 1412–1422 (2001).
Veazey, R.S. et al. Prevention of virus transmission to macaque monkeys by a vaginally applied monoclonal antibody to HIV-1 gp120. Nat. Med. 9, 343–346 (2003).
Meng, G. et al. Primary intestinal epithelial cells selectively transfer R5 HIV-1 to CCR5+ cells. Nat. Med. 2, 150–156 (2002).
Geijtenbeek, T.B.H. et al. DC-SIGN, a dendritic cell-specific HIV-1 binding protein that enhances trans-infection of T cells. Cell 100, 587–597 (2000).
Frank, I. & Pope, M. The enigma of dendritic cell-immunodeficiency virus interplay. Curr. Mol. Med. 2, 229–248 (2002).
Lee, B. et al. cis Expression of DC-SIGN allows for more efficient entry of human and simian immunodeficiency viruses via CD4 and a coreceptor. J. Virol. 75, 12028–12038 (2001).
Turville, S.G. et al. Diversity of receptors binding HIV on dendritic cell subsets. Nat. Immunol. 3, 975–983 (2002).
Spira, A.I. et al. Cellular targets of infection and route of viral dissemination after an intravaginal inoculation of simian immunodeficiency virus into rhesus macaques. J. Exp. Med. 183, 215–225 (1996).
Gupta, P. et al. Memory CD4+ T cells are the earliest detectable human immunodeficiency virus type 1 (HIV-1)-infected cells in the female genital mucosal tissue during HIV-1 transmission in an organ culture system. J. Virol. 76, 9868–9876 (2002).
Collins, K.B., Patterson, B.K., Naus, G.J., Landers, D.V. & Gupta, P. Development of an in vitro organ culture model to study transmission of HIV-1 in the female genital tract. Nat. Med. 6, 475–479 (2000).
Granelli-Piperno, A., Delgado, E., Finkel, V., Paxton, W. & Steinman, R.M. Immature dendritic cells selectively replicate macrophagetropic (M-tropic) human immunodeficiency virus type 1, while mature cells efficiently transmit both M- and T-tropic virus to T cells. J. Virol. 72, 2733–2737 (1998).
Reece, J.C. et al. HIV-1 selection by epidermal dendritic cells during transmission across human skin. J. Exp. Med. 187, 1623–1631 (1998).
McDonald, D. et al. Recruitment of HIV and its receptors to dendritic cell–T cell junctions. Science 300, 1295–1297 (2003).
Pope, M. et al. Conjugates of dendritic cells and memory T lymphocytes from skin facilitate productive infection with HIV-1. Cell 78, 389–398 (1994).
Pope, M., Gezelter, S., Gallo, N., Hoffman, L. & Steinman, R.M. Low levels of HIV-1 infection in cutaneous dendritic cells promote extensive viral replication upon binding to memory CD4+ T cells. J. Exp. Med. 182, 2045–2056 (1995).
Schuitemaker, H. et al. Monocytotropic human immunodeficiency virus 1 (HIV-1) variants detectable in all stages of HIV infection lack T-cell line tropism and syncytium-inducing ability in primary T-cell culture. J. Virol. 65, 356–363 (1991).
Hirsch, V.M. et al. Vpx is required for dissemination and pathogenesis of SIVSM PBj: Evidence of macrophage-dependent viral amplification. Nat. Med. 4, 1401–1408 (1998).
Swingler, S. et al. HIV-1 Nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat. Med. 5, 997–1003 (1999).
Swingler, S. et al. HIV-1 Nef intersects the CD40L signaling pathway in macrophages to promote resting cell infection. Nature (in the press).
Messmer, D. et al. Endogenously expressed nef uncouples cytokine and chemokine production from membrane phenotypic maturation in dendritic cells. J. Immunol. 169, 4172–4182 (2002).
Sol-Foulon, N. et al. HIV-1 Nef-induced upregulation of DC-SIGN in dendritic cells promotes lymphocyte clustering and viral spread. Immunity 16, 145–155 (2002).
Izmailova, E. et al. HIV-1 Tat reprograms immature dendritic cells to express chemoattractants for activated T cells and macrophages. Nat. Med. 9, 191–197 (2003).
Schwartz, E.J. et al. Effect of target cell availability on HIV-1 production in vitro. AIDS 16, 341–345 (2002).
DeBoer, R.J. & Perelson, A.S. Target cell limited and immune control models of HIV infection: a comparison. J. Theor. Biol. 190, 201–214 (1998).
Reilly, C., Schacker, T., Haase, A., Wietgrefe, S. & Krason, D. The clustering of infected SIV cells in lymphatic tissue. J. Am. Stat. Assn. 97, 943–954 (2002).
Reinhart, T.A. et al. Tracking members of the simian immunodeficiency virus deltaB670 quasispecies population in vivo at single-cell resolution. J. Virol. 72, 113–120 (1998).
Veazey, R.S. et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science 280, 427–431 (1998).
Veazey, R.S., Marx, P.A. & Lackner, A.A. Vaginal CD4+ T cells express high levels of CCR5 and are rapidly depleted in simian immunodeficiency virus infection. J. Infect. Dis. 187, 769–776 (2003).
Joag, S.V. et al. Animal model of mucosally transmitted human immunodeficiency virus type 1 disease: intravaginal and oral deposition of simian/human immunodeficiency virus in macaques results in systemic infection, elimination of CD4+ T cells, and AIDS. J. Virol. 71, 4016–4023 (1997).
Mothé, B.R. et al. Dominance of CD8 responses specific for epitopes bound by a single major histocompatibility complex class I molecule during the acute phase of viral infection. J. Virol. 76, 875–884 (2002).
Miller, C.J. et al. Rhesus macaques previously infected with simian/human immunodeficiency virus are protected from vaginal challenge with pathogenic SIVmac239. J. Virol. 71, 1911–1921 (1997).
Amara, R.R. et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292, 69–74 (2001).
Enose, Y. et al. Protection by intranasal immunization of a nef-deleted, nonpathogenic SHIV against intravaginal challenge with a heterologous pathogenic SHIV. Virology 298, 206–316 (2002).
Crotty, S. et al. Protection against simian immunodeficiency virus vaginal challenge by using Sabin poliovirus vectors. J. Virol. 75, 7435–7452 (2001).
Johnson, R.P. et al. Highly attenuated vaccine strains of simian immunodeficiency virus protect against vaginal challenge: inverse relationship of degree of protection with level of attenuation. J. Virol. 73, 4952–4961 (1999).
Joag, S.V. et al. Oral immunization of macaques with attenuated vaccine virus induces protection against vaginally transmitted AIDS. J. Virol. 72, 9069–9078 (1998).
Zhu, T. et al. Persistence of extraordinarily low levels of genetically homogeneous human immunodeficiency virus type 1 in exposed seronegative individuals. J. Virol. 77, 6108–6116 (2003).
Kaul, R. et al. Late seroconversion in HIV-resistant Nairobi prostitutes despite pre-existing HIV-specific CD8+ responses. J. Clin. Invest. 107, 341–349 (2001).
Acknowledgements
We thank C. O'Neill and T. Leonard for help with editing and preparing the manuscript and figures, and J.V. Carlis, T. Schacker and P.J. Southern for comments. M.P. is an Elizabeth Glaser Scientist of the Elizabeth Glaser Pediatric AIDS Foundation and is supported by grants AI52060, AI40877, HD41752 and AI52048 from the National Institutes of Health (NIH), and by the Rockefeller Foundation and the Elizabeth Glaser Pediatric AIDS Foundation. A.T.H. is Regents' Professor and Head of the Department of Microbiology at the University of Minnesota and is supported by grants CA 79458, AI28246, HD37356, AI48484 and RR00167 from the NIH, and by the University of Minnesota.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pope, M., Haase, A. Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat Med 9, 847–852 (2003). https://doi.org/10.1038/nm0703-847
Issue Date:
DOI: https://doi.org/10.1038/nm0703-847
This article is cited by
-
Phagocytosis by an HIV antibody is associated with reduced viremia irrespective of enhanced complement lysis
Nature Communications (2022)
-
The HIV-1 transmission bottleneck
Retrovirology (2017)
-
Broadly neutralizing antibodies suppress post-transcytosis HIV-1 infectivity
Mucosal Immunology (2017)
-
The Effects of Latent Infection on the Dynamics of HIV
Differential Equations and Dynamical Systems (2016)
-
HIV-1 Tat immunization restores immune homeostasis and attacks the HAART-resistant blood HIV DNA: results of a randomized phase II exploratory clinical trial
Retrovirology (2015)
