
Abstract
This review summarizes results from major recent trials regarding novel therapeutic agents in melanoma. The topics discussed include targeted therapy with BRAF (V-RAF murine sarcoma viral oncogene homolog B) inhibitors (vemurafenib and dabrafenib), MEK (mitogen-activated protein kinase kinase) inhibitors (trametinib), bcr-abl/c-kit/PDGF-R inhibitors (imatinib), and angiogenesis inhibitors (bevacizumab and aflibercept), as well as immunotherapy with anti-CTLA-4 (anti-cytotoxic T-lymphocyte antigen-4) antibodies (ipilimumab), anti-PD (anti-programmed death receptor) antibodies (nivolumab and lambrolizumab), and anti-PD-L (anti-programmed death ligand) antibodies. Various combinations of these agents, as well as adjunctive GM-CSF (granulocyte–macrophage colony-stimulating factor), T-VEC (talimogene laherparepvec) oncolytic viruses, and novel chemotherapeutic agents, are also described. Despite the tremendous advances that these novel treatments have created, optimal therapeutic agent selection remains a highly individualized decision. Melanoma therapy has vastly progressed since the days when dacarbazine was the sole option for advanced melanoma patients. The molecular understanding of melanoma pathogenesis has yielded a brighter future for advanced melanoma patients.
Similar content being viewed by others


Novel melanoma therapy includes targeted agents, immunotherapy, and various combinations of agents within these classes |
Unique mechanisms of new therapeutic agents require modified response criteria to properly assess tumor response |
Ongoing studies and numerous clinical trials are investigating combinations of agents that may improve response and survival rates and may delay resistance |
1 Introduction
Although melanoma accounts for less than 5 % of all skin cancers, it is the most deadly because of its propensity for metastatic spread throughout the body. Incidence rates for melanoma in the USA have been increasing over the past 30 years, with an estimated 2013 incidence of 76,690 new melanoma diagnoses and 9,480 deaths. Approximately 1 in 50 persons in the USA will be diagnosed with melanoma in their lifetime [1]. Globally, melanoma was responsible for approximately 232,000 new diagnoses in 2012, with mortality hot spots in Australia and New Zealand [2]. Melanoma staging is organized in accordance with the American Joint Commission on Cancer Tumor–Node–Metastasis (TNM) system. Stage 0 represents intraepithelial disease, stage 1 encompasses localized cutaneous melanoma less than 2 mm thick with no ulceration, and stage 2 includes lesions that are either greater than 2 mm or that are between 1 and 2 mm with ulceration. Regional nodal involvement is indicated by stage 3, and distant metastatic spread is indicated by stage 4 [3]. Five-year relative survival ranges from 89 to 95 % with localized stage 1, from 45 to 77 % with stage 2 melanoma, from 27 to 69 % with regional nodal stage 3 melanoma, and from only 9 to 19 % with distant stage 4 melanoma [4]. Increased understanding of immune regulation and the molecular biology of melanoma have fueled recent treatment advances. This review serves to provide a fundamental understanding of the results of novel treatment strategies that are shaping the prognosis and treatment of metastatic melanoma.
2 Targeted Therapy
2.1 BRAF Inhibition
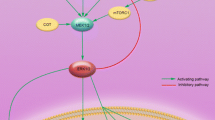
The growth of melanoma involves multiple activating mutations, which promote proto-oncogenes, such as BRAF (V-RAF murine sarcoma viral oncogene homolog B) and KIT (V-KIT Hardy–Zuckerman 4 feline sarcoma viral oncogene homolog) and inhibit tumor suppressors [5]. Approximately 48 % of cutaneous melanomas harbor mutations in the BRAF oncogene [5]. BRAF is a member of the RAF kinase family of growth signal transduction protein kinases, which regulate the MAPK (mitogen-activated protein kinase) pathway (also called the ERK [extracellular signal-regulated kinase] pathway). MAPK/ERK signaling affects cell division, differentiation, and secretion (see Fig. 1) [6, 7]. A series of 197 cases reported by Long et al. demonstrated that the most common BRAF mutation, substitution of glutamic acid at codon 600 for wild-type valine (V600E), accounted for 74 % of all BRAF mutations in metastatic melanomas, followed by the V600K mutation in 20 % [5]. V600R and V600D are additional, less common mutations [8]. Mutated BRAF is constitutively active, fueling the cascade of downstream MEK (MAPK kinase)/ERK enzymes, ultimately resulting in uncontrolled transcription, excessive cell growth, and cancer.

MAPK/ERK (mitogen-activated protein kinase/extracellular signal-regulated kinase) signaling pathway and mechanism of action of mBRAF (mutated V-RAF murine sarcoma viral oncogene homolog B) inhibition. MEK MAPK kinase
Vemurafenib (Zelboraf®) and dabrafenib (Tafinlar®) are small-molecule inhibitors of V600E- and V600K-mutated BRAF kinase. A randomized phase 3 trial (BRIM-3) compared vemurafenib with dacarbazine in patients with previously untreated BRAF V600-mutated metastatic melanoma (see Table 1 for a summary comparison of all discussed trials) [9]. At the first interim analysis, the data safety and monitoring board concluded that both primary endpoints of overall survival and progression-free survival had been met, and they recommended that patients randomized to dacarbazine be allowed to cross over to vemurafenib. In 2011, vemurafenib obtained approval from the US Food and Drug Administration (FDA) for the treatment of V600-mutated melanoma [10]. This agent has since gained approval in the European Union and in several other countries, including Switzerland, Israel, Brazil, New Zealand, and Canada [11]. Updated results of BRIM-3, with a median 12.5 months of follow-up, showed improvements in the median overall survival (13.2 versus 9.6 months; hazard ratio [HR] 0.70, 95 % confidence interval [CI] 0.57–0.87; p < 0.01), progression-free survival (6.9 versus 1.6 months; HR 0.62, 95 % CI 0.49–0.77), and the objective response rate (57.0 versus 8.6 %) [12]. Toxicities associated with vemurafenib therapy included arthralgias (grade 2: 18 %; grade 3: 3 %), rash (grade 2: 10 %; grade 3: 8 %), fatigue (grade 2: 11 %; grade 3: 2 %), nausea (grade 2: 7 %; grade 3: 1 %), alopecia (grade 2: 8 %), and pruritus (grade 2: 6 %; grade 3: 1 %) [9]. At the most recent 12.5-month follow-up, photosensitive skin reactions, including cutaneous squamous cell carcinoma, keratoacanthoma, and skin papilloma, had occurred in 19, 11, and 28 % of patients, respectively [12]. The pathogenesis of vemurafenib’s induction of squamous cell carcinoma and keratoacanthoma involves paradoxical activation of wild-type BRAF in keratinocytes with acquired RAS mutations from prior sun exposure [13].
A second BRAF inhibitor, dabrafenib, gained 2013 approval by the FDA and the European Union on the basis of an open-label phase 3 trial (BREAK-3) of patients with previously untreated BRAF V600-mutated melanoma randomized 3:1 to receive either dabrafenib or dacarbazine (Table 1) [14]. Updated results of the trial included a median progression-free survival of 5.1 months for dabrafenib compared with 2.7 months for dacarbazine, with an HR of 0.30 (95 % CI 0.18–0.53; p < 0.0001) [15]. While the overall survival data were immature, the objective response rate was 53 % for dabrafenib compared with 19 % for dacarbazine. The most frequent adverse events associated with dabrafenib were hyperkeratosis (37 %), headache (32 %), pyrexia (28 %), arthralgia (27 %), and skin papillomas (24 %). Of note, serious adverse events associated with dabrafenib that were life-threatening or resulted in death included pyrexia (4 %), squamous cell carcinoma (6 %), and new primary melanomas (2 %) [15]. The particular toxicity of pyrexia with dabrafenib therapy is poorly understood and is often accompanied by transient elevation of hepatic enzymes, particularly aspartate aminotransferase. There is no correlation between fever and clinical response, and corticosteroids are the only agents to prevent febrile episodes [16]. Other RAF inhibitors are under investigation, such as LGX-818 (a selective BRAF inhibitor) and TAK-632 (a pan-RAF inhibitor) [17, 18].
The remarkable success with BRAF inhibitors has necessitated genetic testing to identify selected patients with BRAF mutations most likely to respond to treatment. A vemurafenib companion diagnostic test, the Cobas® 4800 BRAF Mutation Test, is FDA approved to identify appropriate vemurafenib patients. On the basis of optimal patient outcomes and cost effectiveness, a UK expert panel has recommended BRAF mutation testing of all high-risk patients, ideally activated by the histopathologist prior to referring high-risk patients to the specialist skin cancer multidisciplinary team [19]. This automated molecular assay requires a formalin-fixed and paraffin-embedded melanoma tumor sample, from which DNA is extracted and processed through real-time polymerase chain reaction testing. More than 96 % of V600E BRAF mutations are identified with a standard 125 ng DNA sample containing 5 % mutant alleles, in addition to lower sensitivity for V600K and V600D mutations (requiring greater than 18 % mutant alleles in the tumor sample) [20]. The Cobas® test demonstrates improved accuracy, fewer false negatives, and fewer invalid results, compared with direct sequencing methods [21]. A similar companion real-time polymerase chain reaction test, the THxID® BRAF kit, is available for identification of V600E and V600K BRAF mutations most susceptible to dabrafenib treatment [22]. The identification of BRAF mutations is one of the best examples of a biomarker influencing melanoma treatment response. Other potential biomarkers that are still under investigation include c-kit-mutated and PD-L1 (programmed death ligand-1)-positive melanomas (described below).
Unfortunately, resistance to BRAF inhibition therapy emerges in many patients after approximately 6 months [23]. The proposed mechanisms of resistance to BRAF inhibition, ultimately resulting in reactivation of the MAPK pathway, include upregulation of cancer bypass pathways, such as COT (cancer Osaka thyroid kinase), development of de novo NRAS (neuroblastoma RAS viral (v-ras) oncogene homolog) or MEK mutations, dimerization or variant splicing of BRAF V600E precursor messenger RNA to yield aberrant forms of BRAF V600E, and activation of receptor tyrosine kinases in the BRAF pathway cascade [24].
2.2 MEK Inhibition
In the MAPK/ERK pathway, BRAF is responsible for phosphorylating MEK proteins (MEK 1 and MEK 2), which activate downstream MAPKs (see Fig. 1) [25]. Trametinib (Mekinist™) is a small-molecule inhibitor of MEK 1 and MEK 2, which was approved by the FDA in May 2013 for treatment of BRAF V600E- or V600K-mutated melanoma [26]. A randomized phase 3 trial involving patients with BRAF V600E- or V600K-mutated metastatic melanoma compared trametinib therapy and standard chemotherapy with dacarbazine (Table 1) [27]. Trametinib induced a confirmed complete or partial objective response (according to the Response Evaluation Criteria in Solid Tumors [RECIST]) in 22 %, versus 8 % in the chemotherapy group. Following the study’s primary analysis showing that progression-free survival and overall survival were evidently greater with trametinib therapy, a protocol amendment was added to mandate crossover from dacarbazine to trametinib in patients who experienced disease progression. While the median overall survival results were immature at the time of the data analysis, overall survival at 6 months with trametinib was 81 %, compared with 67 % with dacarbazine (HR 0.65, 95 % CI 0.32–0.92; p = 0.01). The most common adverse events associated with trametinib were rash (57 % for all grades), diarrhea (43 % for all grades), fatigue (26 % for all grades), peripheral edema (26 %), and acneiform dermatitis (19 %). Notably, trametinib treatment was not associated with development of cutaneous neoplasms. This spurred an experimental model investigating MEK inhibitors in combination with BRAF inhibitors, which revealed a reduced incidence of squamous cell carcinoma with the combination regimen [28].
2.3 Combined BRAF and MEK Inhibition
Combined BRAF and MEK inhibition was further investigated in response to the near-universal development of treatment resistance observed with BRAF inhibitor monotherapy. Following the results of an open-label phase 1 and 2 study in 2012, which investigated the combination of dabrafenib and trametinib compared with dabrafenib monotherapy, the FDA granted accelerated approval of the combination regimen in January 2014 (Table 1) [29, 30]. The optimal regimen that improved progression-free survival (9.4 versus 5.8 months) and reduced the hazard of death by 61 % was 150 mg of dabrafenib and 2 mg of trametinib. While the median overall survival data were immature at the time of the analysis, the percentages of patients alive and progression free at 1 year were 41 % in the combination group versus 9 % in the monotherapy group (p < 0.001). Tumors regressed more with the combination regimen (76 versus 54 % with complete or partial response; p = 0.03), which also improved the median duration of response (10.5 versus 5.6 months). Thus, the trametinib plus dabrafenib combination partially overcomes acquired monotherapy treatment resistance.
The incidence of dabrafenib-induced squamous cell carcinoma was reduced from 19 % with dabrafenib monotherapy to 7 % with the combined regimen of dabrafenib 150 mg plus trametinib 2 mg (p = 0.09) [29]. However, the combination regimen was also associated with increased adverse event rates of pyrexia (71 % for all grades; 5 % for grade 3) and chills (58 % for all grades; 2 % for grade 3), as well as pyrexia with severe chills or hypotension, or requiring hospitalization (25 versus 2 %). Several clinical trials are ongoing to further understand the combination of BRAF-MEK inhibition, including a randomized, double-blind, controlled phase 3 trial comparing combined dabrafenib–trametinib therapy and dabrafenib monotherapy; a randomized, open-label, controlled phase 3 trial comparing the combination dabrafenib–trametinib regimen and vemurafenib monotherapy; and a randomized, double-blind, controlled phase 3 trial comparing vemurafenib monotherapy and vemurafenib plus the MEK inhibitor cobimetinib [31–33].
2.4 MEK Inhibition in Uveal Melanoma
The MEK inhibitor selumetinib has recently been studied in uveal melanoma and is the first drug to demonstrate promising results in comparison with traditional chemotherapy. While uveal melanoma is extremely rare, with an incidence of 4.3 cases per million in the USA, it is the most common primary intraocular malignancy in adults [34]. Because of the distinct tumor biology, treatment options for cutaneous melanoma have been virtually ineffective in uveal melanoma. More than 85 % of patients with uveal melanoma harbor mutations in the GNAQ and GNA11 genes, which result in upregulation of the MAPK pathway [35]. This mechanistic knowledge led to a phase 2 investigation of MEK inhibition with selumetinib versus chemotherapy with temozolomide in 80 patients with GNAQ/GNA11-mutated metastatic uveal melanoma (Table 1) [36]. The interim results demonstrated that progression-free survival was 16 weeks with selumetinib versus 4 weeks with chemotherapy, resulting in a 54 % reduction in the risk of progression or death (HR 0.46, 95 % CI 0.30–0.71; p = 0.0005). The overall survival rates were 11.8 months with selumetinib versus 4.7 months with temozolomide, but the difference was not significant (HR 0.79; p = 0.4) [36]. Some degree of tumor shrinkage according to RECIST was observed in 11 % of patients treated with selumetinib versus 0 % of patients treated with chemotherapy. Among patients treated with selumetinib, 28 % experienced grade 3 toxicities (13 % experienced creatine phosphokinase elevation, 8 % liver function test elevation, 3 % rash, 3 % lymphopenia, and 3 % edema) [36].
2.5 Tyrosine Kinase Inhibition
C-kit is a type III transmembrane receptor tyrosine kinase, which binds stem cell factor [37]. Ligand binding induces receptor dimerization, autophosphorylation, and activation of downstream pathways, including MAPK and PI3K (phosphatidylinositol 3-kinase) [37]. Mutations of c-kit are found in 28 % of acral, 39 % of mucosal, and 36 % of chronic sun-damage melanomas, resulting in ligand-independent pathway activation [38]. Imatinib mesylate (Gleevec®) is a small-molecule adenosine triphosphate-competitive inhibitor of several tyrosine kinases, including bcr-abl, c-kit, and PDGF-R, used most notably in the treatment of chronic myelogenous leukemia [39]. A phase 2 study in 2011 investigated imatinib in melanoma patients harboring c-kit mutations or amplifications (Table 1) [40]. Durable responses, defined as partial or complete responses according to RECIST, were reported in 16 % of patients, with a median time to progression of 12 weeks (95 % CI 11–18). The median overall survival was 46.3 weeks in imatinib-treated patients, with greater activity in melanomas that harbored particular recurrent KIT mutations (exon 11 or exon 13) or had mutant alleles in greater abundance than the wild type [40]. The most frequent adverse events associated with imatinib were anemia (61 % for all grades), fatigue (54 % for all grades), nausea (54 % for all grades), periorbital edema (50 % for all grades), and rash (50 % for all grades) [41]. Most melanoma patients that respond to imatinib develop resistance via secondary c-kit mutations [42]. Recent advances in drug sensitivity profiling of melanoma-associated c-kit mutations have highlighted the potential usefulness of a clinical trial to evaluate combined MAPK/PI3K inhibition in imatinib-resistant melanomas [42].
Nilotinib (Tasigna®) is a small-molecule inhibitor from a range of tyrosine kinase inhibitors developed by rational modifications to the imatinib structure, which is FDA approved for the treatment of Philadelphia-positive chronic myelogenous leukemia [43, 44]. An ongoing open-label phase 2 study evaluated nilotinib in 11 patients with metastatic melanoma harboring KIT mutations or amplifications (Table 1) [45]. The preliminary results demonstrated a partial response in 22.2 %, stable disease in 55.6 %, and a decrease in tumor size from baseline in 44.4 %. The two patients with a partial response had KIT mutations and achieved durable responses for 8.4 and 10.0+ months. The most common toxicities associated with nilotinib were grade 1 nausea/vomiting (33.3 %), dry eye (33.3 %), and skin rash (33.3 %), followed by grade 1 headache (22.2 %), grade 1 myalgia (22.2 %), and grade 3 abnormal liver function tests (22.2 %) [45]. As previously mentioned, c-kit mutations are more common in mucosal, acral, and chronic sun damage melanomas. An ongoing phase 2 clinical trial is evaluating the efficacy of nilotinib in c-kit-mutated acral, mucosal, and chronically sun-damaged melanomas that have become either resistant to or intolerant of other tyrosine kinase inhibitors [46].
Another FDA-approved agent for chronic myelogenous leukemia and gastrointestinal stromal tumor has been investigated in melanoma. Developed from an entirely different chemical scaffold from imatinib, dasatinib (Sprycel®) is a small-molecule inhibitor of similar tyrosine kinases, including scr, bcr-abl, c-kit, PDGF-R, and EPHA2 [47, 48]. A single-agent phase 2 trial in 2011 studied dasatinib in an unselected population of 39 patients with stage 3 and 4 previously untreated melanoma (Table 1) [49]. With a response rate of only 5 %, a median progression-free survival of 8 weeks, and a 6-month progression-free survival rate of 13 %, the study failed to meet its primary endpoints. In addition, the chronicity of the most common adverse events of fatigue (83 % for all grades; 14 % for grade 3), dyspnea (75 % for all grades; 6 % for grade 3; 6 % for grade 4), pleural effusion (39 % for all grades; 3 % for grade 3; 6 % for grade 4), nausea (74 % for all grades; 8 % for grade 3), and anorexia (66 % for all grades; 8 % for grade 3) was poorly tolerated in many patients and often necessitated either interruption or dose reduction [49]. The study concluded that single-agent dasatinib had minimal activity and poorly tolerated toxicities in unselected melanoma. However, the potential for identifying a biomarker-selected population remains a viable target. Given the higher prevalence of c-kit mutations in acral and mucosal melanomas, an ongoing phase 2 clinical trial is investigating dasatinib in patients with unresectable, locally advanced or stage 4 mucosal, acral, or vulvovaginal melanomas [50].
2.6 Angiogenesis Inhibition
A final class of targeted melanoma therapy is directed at angiogenesis. Vascular endothelial growth factor (VEGF) is a family of five glycoproteins (VEGF-A, VEGF-B, VEGF-C, VEGF-D, and placental growth factor), which bind three structurally similar tyrosine kinase receptors (VEGFR-1, VEGFR-2, and VEGFR-3) to stimulate blood vessel proliferation and angiogenesis [51]. VEGFR-1 (also known as Flt-1) and VEGFR-2 (also known as Flk-1/KDR) are expressed almost exclusively in endothelial cells, while VEGFR-3 is involved primarily in lymphangiogenesis [51]. In vitro studies have led to the hypothesis that VEGF overexpression in melanoma may induce architectural changes in the tumor vasculature [52]. In addition, VEGF serves as a survival factor for melanoma cells via upregulation of Flk-1/KDR, ERK-1/2, and PI3K signaling [52]. Increased VEGF levels have also been correlated with worse outcomes, particularly in vertical growth melanomas and those with nodal metastases [53].
Bevacizumab (Avastin®) is a humanized monoclonal immunoglobulin G1 antibody against VEGF ligand, which was the first angiogenesis inhibitor approved by the FDA and is commonly used as combination treatment in unresectable, locally advanced, recurrent or metastatic, non-squamous, non-small-cell lung cancer (with carboplatin and paclitaxel); metastatic colon or rectal cancer (with 5-fluorouracil); and metastatic renal carcinoma (with interferon-α) [54]. It should be noted that this novel agent has yet to provide as promising results in melanoma as the other drug classes mentioned in this review. The first major investigation into bevacizumab (in combination with carboplatin and paclitaxel) for melanoma was the 2012, randomized, double-blind, phase 2 BEAM study in 214 patients with previously untreated metastatic melanoma [55]. The trial reported an improvement in the median progression-free survival (5.6 months with versus 4.2 months without bevacizumab; p = 0.1414) and the objective response rate (25.5 versus 16.4 %; p = 0.1577), though these results were not statistically significant. At 17-month follow-up, bevacizumab prolonged the median overall survival (12.3 versus 8.6 months), with a nonsignificant 21 % reduction in the hazard of death (p = 0.1916). The most common toxicities associated with bevacizumab therapy were grade 3 or greater neutropenia (23.8 %), peripheral neuropathy (9.1 %), febrile neutropenia (4.9 %), arterial thromboembolic events (2.15 %), and hypertension (3.5 %). Despite the lack of significant results, the researchers in the BEAM trial believed their findings warranted a phase 3 trial [55]. Possibly because of the recent success of other targeted therapies, such as ipilimumab and vemurafenib, a phase 3 study of first-line bevacizumab in combination with chemotherapy for melanoma has not been pursued to date. An additional phase 2 trial is currently investigating bevacizumab monotherapy compared with dacarbazine, as well as the effectiveness of β-blockers versus angiotensin-converting enzyme inhibitors for the prevention of bevacizumab-induced hypertension [56].
Another investigation of bevacizumab was the randomized, phase 3 AVAST-M trial, which compared bevacizumab therapy and standard observation post-excision as adjuvant treatment following surgical resection for stage 2B, 2C, and 3 cutaneous melanoma (Table 1). The interim results were that bevacizumab significantly improved the disease-free interval rates from 70 to 77 % at 1 year and from 57 to 59 % at 2 years, with a 17 % reduction in the hazard of death (p = 0.03) at a median follow-up interval of 25 months [57]. However, bevacizumab failed to significantly improve the 2-year distant-metastasis-free interval (p = 0.18) or overall survival rates (p = 0.76). Longer follow-up is required to determine the impact on the primary endpoint of 5-year overall survival. Antibody-directed therapy against VEGFR is also being explored. Ramucirumab, a fully humanized monoclonal immunoglobulin G1 antibody against VEGFR-2, is being investigated in a randomized, multicenter phase 2 trial with and without dacarbazine [58].
A final component of anti-angiogenesis therapy is the use of high-affinity soluble decoy VEGFRs. Aflibercept is a fusion protein of the Fc portion of human immunoglobulin G1 with the extracellular ligand binding portions of VEGFR-1 and VEGFR-2. In a single-arm phase 2 study in 2011, aflibercept monotherapy was associated with 50 % progression-free survival of at least 4 months but a limited response rate, with only 7.5 % of patients attaining a partial response (Table 1) [59]. Of interest, the severity of hypertension as an adverse effect of aflibercept was significantly associated with a positive therapy response. Future investigations of aflibercept in combinations and as second- or subsequent-line therapy are warranted.
3 Immunotherapy
3.1 CTLA-4 Blockade
Immune checkpoint blockade consists of antibody therapy targeting immune system components involved in cancer proliferation, such as the T-cell lineage of the adaptive immune system [60]. T-cell activation requires a co-stimulatory signal accomplished by the binding of T-cell CD28 to B7 on the antigen-presenting cell. Cytotoxic T-lymphocyte antigen-4 (CTLA-4) is a protein receptor found on the surface of T-cells, which functions as a natural braking mechanism by binding B7 on the antigen-presenting cell, preventing the required co-stimulatory signal. The resulting T-cell downregulation confers vulnerability to cancer proliferation [61]. Ipilimumab (Yervoy®) is a fully human monoclonal immunoglobulin G1 antibody against CTLA-4, which promotes expansion of T-cell antitumor activity (see Fig. 2). Since achieving FDA approval in 2011 for the treatment of unresectable or metastatic melanoma, ipilimumab has gained approval in several additional countries, including Canada, and in the European Union [62, 63]. A randomized phase 3 trial compared ipilimumab plus dacarbazine therapy and dacarbazine as first-line monotherapy in 502 patients with previously untreated metastatic melanoma (Table 1) [64]. The overall survival of 11.2 months with ipilimumab was a significant improvement over the 9.2 months seen with dacarbazine alone (HR 0.72; p < 0.001). Survival was consistently improved at 1, 2, and 3 years, with a 28 % reduction in the hazard of death and a 24 % reduction in the risk of disease progression with ipilimumab at the 3-year timepoint. Another three-arm, randomized phase 3 trial in 2010 compared the combination of ipilimumab plus a gp100 peptide vaccine, ipilimumab alone, and gp100 peptide vaccine alone as second-line therapy in 676 patients with metastatic melanoma who had undergone previous treatment (Table 1) [65]. The gp100 peptide vaccine was used as an active control in this study, since it induces immune responses [66]. The median overall survival rates were 10.0 months with ipilimumab plus gp100 peptide vaccine, 10.1 months with ipilimumab alone, and 6.4 months with gp100 peptide vaccine alone. The HR of 0.66 with ipilimumab alone compared with gp100 peptide vaccine alone indicated a 34 % reduction in death (p = 0.003), and no differences were found between the two ipilimumab groups (HR 1.04; p = 0.76). The study determined that ipilimumab, either alone or in combination with gp100 peptide vaccine, improved overall survival, compared with gp100 peptide vaccine alone [65].

T-cell activation and mechanism of action of anti-CTLA-4 (anti-cytotoxic T-lymphocyte antigen-4) antibody. MHC major histocompatibility complex, TCR T-cell receptor
Increasing clinical experience with immunotherapeutic agents such as ipilimumab has revealed that traditional RECIST or WHO (World Health Organization) criteria for chemotherapy treatment may not encompass the unique antitumor responses induced by ipilimumab, leading to the development of immune-related response criteria [67]. These modified response criteria acknowledge that shrinkage in baseline lesions, durable stable disease, response after an increase in the total tumor burden, and response in the presence of new lesions may actually represent positive and unique responses to ipilimumab treatment [67]. A major toxicity of ipilimumab’s mechanism of action is immune-related adverse events, which occurred in 58.2 % of patients treated with ipilimumab plus gp100 peptide vaccine and 61.1 % of patients treated with ipilimumab monotherapy, compared with 31.8 % of those treated with gp100 peptide vaccine alone [65]. The immune-related events are predictable and usually mild to moderate but have the potential to be life-threatening. They most commonly affect the skin and gastrointestinal tract, including rash, diarrhea, colitis, and hypophysitis. Algorithms specifying detailed management guidelines have been established [68, 69].
3.2 PD-1 Blockade
PD-1 (programmed death receptor-1) is a fundamental immune-checkpoint receptor expressed on activated T cells and has proven to be an exciting target in melanoma immunotherapeutics. PD-1 has two ligands, PD-L1 and PD-L2, which are members of the B7 protein family and are expressed on both tumor and stromal cells [70]. PD-1 ligand binding results in T-cell exhaustion, inhibition, and immunosuppression [70]. Nivolumab is a fully human immunoglobulin G4 monoclonal antibody directed against PD-1 and promotes antitumor activity by affecting T-cell activation downstream from ipilimumab (see Fig. 3) [71]. Long-term follow-up results from a 2012 phase 1 study of stage 4 melanoma patients treated with nivolumab revealed durable objective responses in 31 % of patients (Table 1) [72, 73]. Overall survival was 62 % at 1 year, 44 % at 2 years, and 40 % at 3 years, with a median overall survival of 16.8 months across all doses. This study also analyzed tumor specimens from patients with melanoma, non-small-cell lung cancer, colorectal cancer, and renal cancer for the expression of PD-L1. Adverse events related to nivolumab therapy included grade 3–4 lymphopenia (3 %), fatigue, increased lipase (2 %), diarrhea (2 %), endocrine disorders (2 %), and hepatitis (1 %). An objective response was observed in 36 % of PD-L1-positive tumors but in 0 % of PD-L1-negative tumors [72]. However, these results require cautionary interpretation, since the tumor samples were optional and the methodology to evaluate PD-L1 is under development. The utility of PD-L1 as a biomarker of response with anti-PD-1 blockade is appealing but warrants further investigation [72].

PD-1 (programmed death receptor-1) pathway and mechanism of action of anti-PD-1 and anti-PD-L1 (anti-programmed death ligand-1) antibodies. MHC major histocompatibility complex, TCR T-cell receptor
Another investigation into anti-PD-1 therapy involves the humanized monoclonal immunoglobulin G4 antibody lambrolizumab. In an ongoing, open-label phase 1B trial, three dosing regimens of lambrolizumab were administered to patients with advanced melanoma: 10 mg/kg every 2 weeks, 10 mg/kg every 3 weeks, or 2 mg/kg every 3 weeks (Table 1) [74]. The overall confirmed response rate according to RECIST for all three cohorts was 38 %, with the highest response in the cohort receiving 10 mg/kg every 2 weeks (52 %). The study found that median progression-free survival was greater than 7 months, and 81 % of patients who had responded to treatment continued to respond. Among patients who had past exposure to ipilimumab therapy, the 38 % response rate to lambrolizumab was similar to the 37 % response rate in patients with no prior exposure to ipilimumab. Thus, patients who fail one type of checkpoint inhibition may still respond to another class of antibody therapy [74]. A phase 2 trial is investigating lambrolizumab therapy in advanced melanoma patients who have received prior treatment, including ipilimumab [75]. Preliminary results of an ongoing phase 1 trial with the engineered PD-L1 antibody MPDL3280A in patients with locally advanced or metastatic melanoma included an overall response rate of 26 %, with 6-month progression-free survival of 35 % [76].
3.3 Combined CTLA-4 and PD-1 Blockade
The combination of CTLA-4 and PD-1 antibody blockade with ipilimumab and nivolumab, respectively, has been studied [77]. This phase 1 trial examined cohorts of patients receiving concurrent intravenous ipilimumab and nivolumab every 3 weeks for four doses, followed by nivolumab alone every 3 weeks for four doses (Table 1). The study also included a sequenced regimen of patients previously treated with ipilimumab who then received nivolumab monotherapy every 2 weeks for up to 48 doses. Among the 53 patients receiving the concurrent regimen, an objective response according to the modified WHO criteria was observed in 40 % (21 of 52 patients). Tumor reduction greater than 80 % was observed in 16 of these patients at 12 weeks, with a complete response observed in five patients [77]. While the results of ipilimumab plus nivolumab combination therapy are striking, it is difficult to compare them with previous studies of monotherapy with either agent, because of the use of different response criteria (WHO versus RECIST). In the sequenced regimen, 20 % of patients had an objective response, with 13 % achieving tumor reductions of 80 % or more at 8 weeks. Thus, patients previously treated with CTLA-4 blockade may continue to respond to PD-1 blockade with nivolumab. Serious adverse events related to the concurrent regimen occurred in 49 % of patients; hepatic events (15 %), gastrointestinal events (9 %), and renal events (6 %) were the most common grade 3 or 4 toxicities. A phase 3 study is comparing nivolumab monotherapy, ipilimumab monotherapy, and nivolumab plus ipilimumab combination therapy [78].
3.4 Combined CTLA-4 Blockade and BRAF Inhibition
The success of BRAF inhibition with vemurafenib and CTLA-4 blockade with ipilimumab has spurred investigation into the combination of these two drug classes for the treatment of metastatic melanoma. The differing mechanisms of action for vemurafenib and ipilimumab were postulated to enhance clinical benefit over single agents alone [79]. A phase 1 study examined concurrent vemurafenib and ipilimumab treatment in metastatic melanoma patients with a BRAF V600 mutation [80]. Two cohorts of patients were enrolled, the first receiving vemurafenib and ipilimumab each at their full approved dose (starting with vemurafenib alone for 1 month and then concurrent treatment with both drugs), and the second receiving a lower dose of vemurafenib concurrently with full-dose ipilimumab. Grade 3 elevations in aminotransferase levels occurred in 80 % of patients in cohort 1 and in 33.3 % of patients in cohort 2, while grade 2 elevations in hepatic enzymes occurred in 16.7 % of patients in cohort 2. While these adverse events were reversible with glucocorticoid administration or discontinuation of the drugs, the liver toxicity ultimately resulted in early termination of the study [80]. To date, there have been no successful combinations of targeted therapy with immunotherapy. Clearly, while inter-class drug combinations are appealing, strategies must be carefully considered and studied.
3.5 Combined CTLA-4 Blockade and GM-CSF
A recent study has explored the use of ipilimumab with granulocyte–macrophage colony-stimulating factor (GM-CSF), a cytokine that simulates white blood cell development from stem cells [81]. While GM-CSF has traditionally been used to treat neutropenia, recent studies have explored its use as an immune adjuvant for cancer treatment to improve efficacy and safety. A randomized phase 2 trial examined the addition of GM-CSF to ipilimumab, compared with ipilimumab monotherapy, in patients with metastatic melanoma (Table 1) [82]. At 1 year, overall survival was 68.9 % with the combination regimen versus 52.9 % with ipilimumab monotherapy (p = 0.014), while tumor shrinkage rates at median follow-up of 13.3 months were comparable (11.3 % with combination therapy versus 14.7 % with monotherapy). At the time of analysis, median overall survival had not been reached for the ipilimumab plus GM-CSF combination regimen but was 12.6 months with ipilimumab alone. The addition of GM-CSF to ipilimumab was associated with less serious adverse events, with grade 3–5 adverse events occurring in 45 % of patients compared with 58 % of patients receiving ipilimumab monotherapy (p = 0.038). In particular, grade 5 toxicities, such as colonic perforation, multi-organ failure, hepatic failure, and respiratory failure, were more common with ipilimumab monotherapy [82]. Further studies will explore the addition of GM-CSF to ipilimumab [83].
3.6 T-VEC
Talimogene laherparepvec, or T-VEC for short, is an engineered herpes simplex virus-1 designed to target and replicate within cancer cells, leading to cell lysis and an immune reaction against the cancer [84]. T-VEC virus modifications include deletion of the ICP34.5 gene, which ordinarily allows the virus to infect healthy neurons, and deletion of the ICP47 gene, which ordinarily shields the virus from the immune system [84]. In addition, the oncolytic virus is engineered with insertion of the human GM-CSF gene. With a particular predilection for cancer cells, T-VEC replicates within tumors, secreting GM-CSF and leading to cancer cell lysis. The release of tumor-specific antigens and attraction of dendritic cells by GM-CSF leads to activation of the patient’s adaptive immune system [85]. A phase 2 trial in 2009 reported a 26 % objective response rate according to RECIST, 1-year overall survival of 58 %, and 2-year overall survival of 52 % (Table 1) [86]. The randomized, open-label, phase 3 OPTiM study compared T-VEC with GM-CSF in patients with melanoma and regional or distant metastases [87]. The primary endpoint of the study was the durable response rate, defined as a partial or complete response lasting at least 6 months, starting within 12 months of treatment. The durable response rate was 16 % in patients receiving T-VEC, compared with 2 % in those receiving GM-CSF (p < 0.0001). The objective response rate, which included any positive tumor response, was 26 % with T-VEC, including complete response in 11 % of patients, compared with an objective response rate of 6 % and only a 1 % complete response rate with GM-CSF. The most common adverse events with T-VEC were fatigue, chills, and pyrexia. A planned interim analysis demonstrated a trend toward improved overall survival, with a 21 % reduction in the hazard of death [87].
4 Additional Novel Treatment Results
While the major focus of this review is on novel targeted and immunotherapies, a brief mention will be made regarding advances with a chemotherapeutic agent. Nab-paclitaxel is a chemotherapeutic albumin-bound agent, which utilizes the water-soluble properties of albumin to bind to the tumor, inhibit mitosis, and ultimately induce tumor apoptosis [88]. While this anticancer compound is FDA approved for metastatic breast cancer, non-small-cell lung cancer, and metastatic pancreatic adenocarcinoma, it has demonstrated significant activity in melanoma as well. A randomized, open-label phase 3 study compared nab-paclitaxel and dacarbazine in chemotherapy-naive patients with metastatic melanoma (Table 1). Nab-paclitaxel demonstrated a significantly improved progression-free survival of 5.4 months versus 2.5 months with dacarbazine (HR 0.715, p = 0.088) [89]. The interim analysis also demonstrated nonsignificant trends toward an improved median overall survival (12.7 months versus 11.1 months; HR 0.845, p = 0.330) and an improved response rate (15 versus 11 %; p = 0.239). An interim subanalysis determined that while the treatment benefit of nab-paclitaxel versus dacarbazine was greatest in wild-type BRAF (p = 0.088), the benefit trend was observed in all patients regardless of their BRAF mutation status. Adverse events observed in greater than 10 % of nab-paclitaxel patients were neuropathy (25 versus 0 %; p < 0.01) and neutropenia (20 versus 10 %; p = 0.004) [88].
5 Conclusion
This review serves as an overview of novel therapeutic investigations for melanoma patients. Both targeted therapy and immunotherapy have revolutionized prognostic outcomes in advanced melanoma disease. It must be emphasized, however, that this field is rapidly evolving and changing. This review attempts to present the most current interim results and highlight selected ongoing clinical trials and future investigations. FDA approval of many agents discussed in this review has created opportunities for highly individualized selection of an optimal therapeutic agent. Increasingly available genetic testing to determine biomarkers that predict treatment response allows for identification of cohorts most likely to benefit from a particular treatment. Melanoma therapy has vastly progressed since the days when dacarbazine was the sole option for advanced melanoma patients. Exploration and understanding of the molecular pathogenesis of melanoma has yielded a brighter future for patients and their families battling advanced melanoma.
References
Rigel DS, Russak J, Friedman R. The evolution of melanoma diagnosis: 25 years beyond the ABCDs. CA Cancer J Clin. 2010;60(5):301–16.
Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0, cancer incidence and mortality worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer; 2013. http://globocan.iarc.fr. Accessed 3 Feb 2014.
Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199.
Balch CM, Buzaid AC, Soong SJ, et al. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J Clin Oncol. 2001;19(16):3635–48.
Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–46.
Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–47.
Ribas A, Flaherty KT. BRAF targeted therapy changes the treatment paradigm in melanoma. Nat Rev Clin Oncol. 2011;8(7):426–33.
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–67.
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
US Food and Drug Administration. FDA news release: FDA approves Zelboraf and companion diagnostic test for late-stage skin cancer. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm268241.htm. Accessed 9 Feb 2014.
Onco’Zine. First personalized cancer medicine allows patients with deadly form of metastatic melanoma to live significantly longer. http://oncozine.com/profiles/blogs/first-personalized-cancer-medicine-allows-patients-deadly-form-of. Accessed 9 Feb 2014.
Chapman PB, Hauschild A, Robert C, et al. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (VEM) with dacarbazine (DTIC) in previously untreated patients with BRAF V600E-mutated melanoma. J Clin Oncol, ASCO Annual Meeting Abstract 2012;30(suppl):8502.
Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF. N Engl J Med. 2012;366(3):207–15.
Hauschild A, Grob J, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65.
Hauschild A, Grob JJ, Demidov LV, et al. Phase III, randomized, open-label, multicenter trial (BREAK-3) comparing the BRAF kinase inhibitor dabrafenib (GSK2118436) with dacarbazine (DTIC) in patients with BRAF V600E-mutated melanoma. J Clin Oncol, ASCO Annual Meeting Abstract 2012;30(suppl):LBA8500.
Lee CI, Menzies AM, Haydu L, et al. Correlates of fever in patients (pts) receiving combined dabrafenib (GSK2118436) plus trametinib (GSK1120212) for V600 BRAF-mutant metastatic melanoma (MM). J Clin Oncol, ASCO Annual Meeting Abstracts 2012;30(suppl):E19011.
Kefford R, Miller WH, Shao-Weng D, et al. Preliminary results from a phase Ib/II, open-label, dose-escalation study of the oral BRAF inhibitor LGX818 in combination with the oral MEK 1/2 inhibitor MEK162 in BRAFV600-dependent advanced solid tumors. J Clin Oncol, ASCO Annual Meeting Abstract 2013;31(suppl):9029.
Nakamura A, Arita T, Tsuchiya S, et al. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013;73(23):7043–55.
Gonzalez D, Fearfield L, Nathan P, et al. BRAF mutation testing algorithm for vemurafenib treatment in melanoma: recommendations from an expert panel. Br J Dermatol. 2013;168(4):700–7.
Halait H, Demartin K, Shah S, et al. Analytical performance of a real-time PCR-based assay for V600 mutations in the BRAF gene, used as the companion diagnostic test for the novel BRAF inhibitor vemurafenib in metastatic melanoma. Diagn Mol Pathol. 2012;21(1):1–8.
Cobas® 4800 BRAF V600 Mutation Test Package Insert. Roche Molecular Systems, Inc. August 2011.
US Food and Drug Administration. THxID™-BRAF kit for use on the ABI 7500 Fast Dx Real-Time PCR Instrument—P120014. BioMérieux labeling, May 2013. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cftopic/pma/pma.cfm?num=p120014. Accessed 2 Mar 2014.
Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–14.
Chapman PB. Mechanisms of resistance to RAF inhibition in melanomas harboring a BRAF mutation. Am Soc Clin Oncol Educ Book 2013: 80–2.
Qi M, Elion EA. MAP kinase pathways. J Cell Sci. 2005;118(Pt 16):3569–72.
US Food and Drug Administration. Trametinib. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm354478.htm. Accessed 12 Feb 2014.
Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–14.
Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–15.
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703.
US Food and Drug Administration. Trametinib and dabrafenib. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm381451.htm. Accessed 17 Feb 2014.
GlaxoSmithKline. A study comparing trametinib and dabrafenib combination therapy to dabrafenib monotherapy in subjects with BRAF-mutant melanoma [ClinicalTrials.gov identifier NCT01584648]. US National Institutes of Health, ClinicalTrials.gov [online]. http://clinicaltrials.gov/show/NCT01584648. Accessed 20 Aug 2013.
GlaxoSmithKline. Dabrafenib plus trametinib vs vemurafenib alone in unresectable or metastatic BRAF V600E/K cutaneous melanoma (COMBI-v) [ClinicalTrials.gov identifier NCT01597908]. http://clinicaltrials.gov/show/NCT01597908. Accessed 20 Aug 2013.
Hoffman La Roche. A phase 3 study comparing GDC-0973, a MEK inhibitor, in combination with vemurafenib vs vemurafenib alone in patients with metastatic melanoma [ClinicalTrials.gov identifier NCT01689519]. http://clinicaltrials.gov/show/NCT01689519. Accessed 25 Aug 2013.
Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973–1997. Ophthalmology. 2003;110(5):956–61.
Van Raamsdonk CD, Griewank KG, Crosby MB, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363(23):2191–9.
Carvajal RD, Sosman JA, Quevedo F, et al. Phase II study of selumetinib (sel) versus temozolomide (TMZ) in gnaq/Gna11 (Gq/11) mutant (mut) uveal melanoma. J Clin Oncol, ASCO Annual Meeting Abstract 2013;31(suppl):9003.
Alexeev V, Yoon K. Distinctive role of the cKit receptor tyrosine kinase signaling in mammalian melanocytes. J Invest Dermatol. 2006;126(5):1102–10.
Ko JM, Velez NF, Tsao H. Pathways to melanoma. Semin Cutan Med Surg. 2010;29(4):210–7.
Guilhot F. Indications for imatinib mesylate therapy and clinical management. Oncologist. 2004;9(3):271–81.
Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327–34.
Carvajal RD, Antonescu CR, Wolchok JD, et al. Supplementary appendix to: KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327–34.
Todd JR, Becker TM, Kefford RF, et al. Secondary c-kit mutations confer acquired resistance to RTK inhibitors in c-kit mutant melanoma cells. Pigment Cell Melanoma Res. 2013;26(4):518–26.
Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant BCR-ABL. Cancer Cell. 2005;7(2):129–41.
US Food and Drug Administration. Nilotinib (Tasigna). http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/ucm216218.htm. Accessed 12 Feb 2014.
Cho JH, Kim KM, Kwon M, et al. Nilotinib in patients with metastatic melanoma harboring kit gene aberration. Invest New Drugs. 2012;30(5):2008–14.
Dana-Farber Cancer Institute; Brigham and Women’s Hospital; Beth Israel Deaconess Medical Center; Massachusetts General Hospital; Novartis. Nilotinib in TKI resistant or intolerant patients with metastatic mucosal, acral, or chronically sun damaged melanoma [ClinicalTrials.gov identifier NCT00788775]. http://clinicaltrials.gov/show/NCT00788775. Accessed 25 Aug 2013.
Lombardo LJ, Lee FY, Chen P, et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658–61.
Shah NP, Tran C, Lee FY, et al. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305(5682):399–401.
Kluger HM, Dudek AZ, McCann C, et al. A phase 2 trial of dasatinib in advanced melanomas. Cancer. 2011;117(10):2202–8.
Eastern Cooperative Oncology Group; National Cancer Institute. Dasatinib in treating patients with locally advanced or metastatic mucosal melanoma, acral melanoma, or vulvovaginal melanoma that cannot be removed by surgery [ClinicalTrials.gov identifier NCT00700882]. http://clinicaltrials.gov/show/NCT00700882. Accessed 28 Aug 2014.
Corrie PG, Basu B, Zaki KA. Targeting angiogenesis in melanoma: prospects for the future. Ther Adv Med Oncol. 2010;2(6):367–80.
Graells J, Vinyals A, Figueras A, et al. Overproduction of VEGF concomitantly expressed with its receptors promotes growth and survival of melanoma cells through MAPK and PI3K signaling. J Invest Dermatol. 2004;123(6):1151–61.
Goydos JS, Gorski DH. Vascular endothelial growth factor CmRNA expression correlates with stage of progression in patients with melanoma. Clin Cancer Res. 2003;9(16 Pt 1):5962–7.
National Cancer Institute. FDA approval for bevacizumab. http://www.cancer.gov/cancertopics/druginfo/fda-bevacizumab. Accessed 31 Jul 2013.
Kim KB, Sosman JA, Fruehauf JP, et al. BEAM: a randomized phase II study evaluating the activity of bevacizumab in combination with carboplatin plus paclitaxel in patients with previously untreated advanced melanoma. J Clin Oncol. 2012;30(1):34–41.
Haukeland University Hospital, The Norwegian Melanoma Group and the Norwegian Cancer Association. Bevacizumab versus dacarbazine in metastatic melanoma [ClinicalTrials.gov identifier NCT01705392]. http://clinicaltrials.gov/show/NCT01705392. Accessed 28 Aug 2013.
Corrie P, Marshall A, Goonewardena M, et al. Adjuvant bevacizumab as treatment for melanoma patients at high risk of recurrence: preplanned interim results for the AVAST-M trial. J Clin Oncol, ASCO Annual Meeting Abstract 2013;31(suppl):9000.
ImClone LLC. A study of ramucirumab with or without dacarbazine in metastatic malignant melanoma [ClinicalTrials.gov identifier NCT00533702]. http://clinicaltrials.gov/show/NCT00533702. Accessed 25 Aug 2013.
Tarhini AA, Frankel P, Margolin KA, et al. Aflibercept (VEGF Trap) in inoperable stage III or stage IV melanoma of cutaneous or uveal origin. Clin Cancer Res. 2011;17(20):6574–81.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619.
Lee B, Mukhi N, Liu D. Current management and novel agents for malignant melanoma. J Hematol Oncol. 2012;5(3):1–7.
US Food and Drug Administration. FDA approves new treatment for a type of late-stage skin cancer. http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm1193237.htm. Accessed 12 Feb 2014.
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–26.
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15.
Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–20.
Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691–7.
Chin K, Ibrahim R, Berman D, et al. Treatment guidelines for the management of immune-related adverse events in patients treated with ipilimumab, and anti-CTLA4 therapy. Ann Oncol. 2008;19(8 Suppl):viii244–5.
Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24(2):207–12.
Ribas A. Tumor immunotherapy directed at PD-1. NEJM. 2012;366(26):2517–9.
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Sznol M, Kluger HM, Hodi FS, et al. Survival and long-term follow-up of safety and response in patients (pts) with advanced melanoma (MEL) in a phase I trial of nivolumab (anti-PD-1; BMS-936558; ONO-4538). J Clin Oncol, ASCO Annual Meeting Abstract 2013;31(suppl):CRA9006.
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (Anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44.
Merck. Study of MK-3475 (lambrolizumab) in participants with progressive locally advanced or metastatic carcinoma, melanoma, or non-small cell lung carcinoma (P07990/MK-3475-001 AM7) [ClinicalTrials.gov identifier NCT01295827]. http://clinicaltrials.gov/show/NCT01295827. Accessed 20 Aug 2013.
Hamid O, Sosman JA, Lawrence DP, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma. J Clin Oncol, ASCO Annual Meeting Abstract 2013;31(suppl):CRA9010.
Wolchock JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–33.
Bristol-Myers Squibb. Phase 3 study of nivolumab or nivolumab plus ipilimumab versus ipilimumab alone in previously untreated advanced melanoma [ClinicalTrials.gov identifier NCT01844505]. http://clinicaltrials.gov/show/NCT01844505. Accessed 20 Aug 2013.
Liu C, Peng W, Xu C, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19(2):393–403.
Ribas A, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–6.
Shi Y, Liu CH, Roberts AI, et al. Granulocyte–macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res. 2006;16(2):126–33.
Hodi FS, Lee SJ, McDermott DF, et al. Multicenter, randomized phase II trial of GM-CSF (GM) plus ipilimumab (Ipi) versus Ipi alone in metastatic melanoma: E1608. J Clin Oncol, ASCO Annual Meeting Abstract 2013;31(suppl):9007.
The ASCO Post. ASCO 2013: adding GM-CSF to ipilimumab significantly improves survival for patients with metastatic melanoma. http://www.ascopost.com/ViewNews.aspx?nid=4188. Accessed 1 Feb 2014.
Liu BL, Robinson M, Han Z-Q, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303.
Kaufman HL, Kim DW, DeRaffele G, et al. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIC and IV melanoma. Ann Surg Oncol. 2010;17(3):718–30.
Senzer NN, Kaufman HL, Amatruda T, et al. Phase II clinical trial of a granulocyte–macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patents with unresectable metastatic melanoma. J Clin Oncol. 2009;27(334):5763–71.
Andtbacka RHI, Collichio FA, Amatruda T, et al. OPTiM: a randomized phase iii trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte–macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol, ASCO Annual Meeting Abstract 2013;31(suppl):9008.
Hersh E, Del Vecchio M, Brown M, et al. Phase 3, randomized, open-label, multicenter trial of nab-paclitaxel (nab-P) versus dacarbazine (DTIC) in previously untreated patients with metastatic malignant melanoma [abstract]. Pigment Cell Melanoma Res. 2012;25(6):863–903.
Hersh E, Del Vecchio M, Brown MP, et al. A phase III trial of nab-paclitaxel versus dacarbazine in chemotherapy-naive patients with metastatic melanoma: a subanalysis based on BRAF status. J Clin Oncol, ASCO Annual Meeting Abstract 2013;31(suppl):9030.
Acknowledgments
The authors would like to express their thanks to Chanel Karimkhani, who designed the figures included in this article No sources of funding were used in the preparation of this article, and the authors have no financial or other conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Karimkhani, C., Gonzalez, R. & Dellavalle, R.P. A Review of Novel Therapies for Melanoma. Am J Clin Dermatol 15, 323–337 (2014). https://doi.org/10.1007/s40257-014-0083-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-014-0083-7