
Abstract
Since the end of 2019, a new coronavirus strain has been reported in the Chinese province of Wuhan, indicated as 2019-nCoV or SARS-CoV-2. In February 2020, the first case of transmission on Italian soil was reported. On March 09, 2020, at the time of protocol design, the Italian Ministry of Health reported 10,149 people who had contracted the virus; of these, 8514 were positive, of which 5038 were hospitalized with symptoms (59.2%) and 877 in intensive care (10.3%), while the remaining 2599 were in home isolation; 631 were deceased (6.2%) and 1004 healed (9.9%). To date there are no studies in the literature that demonstrate its feasibility and efficacy in the context of the worldwide SARS-CoV-2 epidemic. Based upon the little existing evidence, we planned to assess the efficacy of the infusion of hyperimmune plasma in COVID-19 patients in a one-arm proof-of-concept clinical trial. The primary objective of our study is to evaluate the efficacy of the administration of plasma taken from convalescent donors of COVID-19 to critically ill patients with COVID-19 in terms of their survival. Death from any cause will be considered. The main limit of this study is its one-arm proof-of-concept design with only 43 patients enrolled. However, in the absence of previous evidence, larger and/or randomized trials did not appear to be ethically acceptable. Moreover, the results from this study, if encouraging, will allow us to plan further informed large clinical trials. Trial registration: NCT 04321421 March 23, 2020.
Similar content being viewed by others

Background and rationale
Since the end of 2019, a new coronavirus strain has been reported in the Chinese province of Wuhan, indicated as 2019-nCoV or SARS-CoV-2, responsible for a syndrome called new coronavirus disease 2019 (COVID-2019, Coronavirus 2019 Infected Disease) whose clinical picture includes flu-like symptoms, up to pneumonia (SARS-CoV-2 pneumonia) and in extreme cases up to severe acute respiratory failure which may require intensive care [1,2,3]. Already at the beginning of 2020, the first cases were registered also on national territory and in February 2020, the first case of transmission on Italian soil was reported. On March 09, 2020, at the time of protocol design, the Italian Ministry of Health reported 10,149 people who had contracted the virus; of these, 8514 were positive, of which 5038 were hospitalized with symptoms (59.2%) and 877 in intensive care (10.3%), while the remaining 2599 were in home isolation; 631 were deceased (6.2%) and 1004 healed (9.9%).
There is currently no specific treatment for SARS-CoV-2. Passive immunization for the prevention and treatment of human infectious diseases and the related concept of artificially acquired passive immunity dates back to the first experiences of infection with the hepatitis B virus, avian, and more recently with Ebola virus. The possibility of immediately obtaining immunization against infectious agents by administering specific antibodies to the pathogen contained in the plasma obtained from the recovered/convalescent subjects has proven possible efficacy in patients without a therapeutic alternative, i.e. when specific vaccines or other pharmaceutical treatments are not available. Initial experiences using hyperimmune plasma in China were published in the international literature [4,5,6,7].
As for other previous viral epidemics, such as Ebola, MERS-CoV (Middle East Respiratory Syndrome coronavirus, MERS-CoV) and H1N1pdm09 (pandemic 2009 influenza A H1N1), the use of plasma from convalescent subjects, survivors of the infection, can have a therapeutic role, above all because it is feasible and not very risky. The clinical data on such treatment are few and mainly derive from case reports [4,5,6, 8,9,10,11,12,13]. However, the analysis of the literature to identify possible benefits deriving from this treatment on patients suffering from other viral infections can be useful to help set up a better clinical management of COVID-19. To this end, a meta-analysis was carried out in 2015 to evaluate the clinical efficacy of the administration of plasma or serum for the treatment of acute respiratory infections with viral aetiology to be able to manage clinically the MERS-CoV infection [11]. Although the studies analysed were considered of low quality, without control groups and with moderate or high risk of bias, the authors concluded that the administration of plasma or serum from immunized patients could be applied safely, recording a reduction in hospital stay and above all lower mortality rate. A possible explanation of the effectiveness of convalescent plasma is the suppression of viraemia by antibodies contained in the plasma. Generally, the viral load peaks in the first week of infection and the patient develops a primary immune response within 10–14 days, followed by virus clearance. For this reason, it may be more effective to administer convalescent plasma in the early stage of the disease.
Considering the evidence from the literature review, the use of plasma from convalescent donors could have a therapeutic role, without serious adverse events in critically ill patients with COVID-19. Furthermore, the possibility of having local donors offers the added value of giving a specific immunity acquired against the infectious agent of the local strain, in consideration of the fact that in other areas the strain may be different. The possibility of collecting plasma by plasmapheresis procedure quickly and effectively, making it immediately available to the patient who needs it, represents at this moment a further therapeutic possibility. To date, however, there are no studies in the literature that demonstrate its feasibility and efficacy in the context of the worldwide SARS-CoV-2 epidemic [14]. Based on the little existing evidence, we planned to assess the efficacy of the infusion of hyperimmune plasma in COVID-19 patients.
Methods
Study objectives and endpoints
The primary objective of our study is to evaluate the efficacy of the administration of plasma taken from convalescent donors of COVID-19 to critically ill patients with COVID-19 in terms of their survival. Death from any cause will be considered.
The secondary objective is to evaluate the efficacy in terms of clinical outcomes, assessed as
-
1.
Time to extubation.
-
2.
Length of intensive care stay.
-
3.
Days of mechanical ventilation.
-
4.
Time to CPAP weaning.
-
5.
Viral load.
-
6.
Immune response.
Trial design and participants
This is a multicentre one-arm proof-of-concept interventional study. This study will be conducted at the COVID units of the participating centers in accordance with consensus ethical principles derived from the Declaration of Helsinki, as well as the quality standards of Good Clinical Practice. This study has been registered at clinicaltrials.gov as NCT 04321421. It has been approved by the Local Ethical Committee on March 17th, 2020.
The study is expected to last from March to May 2020. Each patient will be followed up for 7 days since enrolment.
Recruitment and selection of convalescent donors
The convalescent patients will be recruited according to a call list established by the specialists of the Infectious Diseases Unit of the participating institutions. Donors will be male, aged 18 years or older, who had recovered from COVID-19 disease (defined as 2 consecutive negative naso-pharyngeal swabs) since not less than 7 days and not more than 30 days. The donors will be registered according to the national regulation and thoroughly clinically evaluated by the local physician, with the purpose of highlighting any absolute contraindications to the aphaeresis procedure. All donors will need to test negative for hepatitis A and E RNA, and parvovirus 19 DNA, as well as for hepatitis B, C, HIV and syphilis in the molecular test (according to the current law).
Plasma collection from the selected donors and validation procedures
The plasma collection will be performed in a dedicated facility, using the latest generation cell separator device, set according to the donor characteristics, under nurses’ supervision. We will collect a volume of plasma ranging from 500 to 600 ml. We will administer calcium gluconate for prophylaxis of citrate dextrose acid formula A-induced hypo-calcemia, as per routine. Collected plasma will need to show a neutralizing title of 1:160 or more.
A sample will be sent to the Virology laboratory to perform the neutralization test for the identification of anti-SARS-CoV-2-neutralizing antibodies. Briefly serum samples will be titrated in a fourfold dilution series in 96-well tissue culture microtiter plate and mixed with a working dilution of a SARS-CoV-2 strain. After 1 h incubation at 33 °C and 5% CO2, VERO E6 cells will be added. At 48 h of incubation at 33 °C and 5% CO2, wells will be stained with Gram’s crystal violet solution for 30 min. Microtiter plates will be then washed in running water. Wells will be scored to evaluate the degree of cytopathic effect (CPE) compared to the virus control. Blue staining of wells indicates the presence of neutralizing antibodies. Neutralizing titre is the maximum dilution with the reduction of 90% of CPE. A positive titre is equal to or greater than 1/10.
Immediately after the collection, each plasma unit will be divided into two sub-units of about 250–300 ml, using a sterile connector, labelled as hyperimmune COVID plasma and stored in a dedicated freezer, at a controlled temperature ranging from − 40 to − 25 °C. As per routine, the plasma will be validated and made available for infusion at the completion of all tests. Pathogen reduction of plasma will also be performed, as specifically required by the National Centre for Blood in the framework of the trial.
The transfer of fresh or frozen plasma to the clinical units for administration to the patients enrolled in the trial will occur upon request by the treating physician using the established local procedures, inclusive of electronic tracking and ABO transfusion compatibility. The dose of plasma to be infused will be decided based on the clinical condition of each patient.
Eligibility criteria for COVID patients
Compromised patients with a COVID-19 infection hospitalized in the COVID units of the participating centers will be enrolled. The full eligibility criteria are detailed in Table 1
Study intervention and procedures
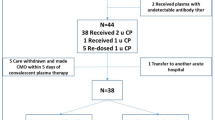
Upon receipt of the plasma at the requesting clinical unit, the 250/300 ml plasma unit is infused immediately to the patient (day 1), with appropriate clinical and instrumental monitoring, as per current clinical practice. In the absence of clinical response (O2 saturation) within 48 h (day 3), a second dose may be administered; a third unit may be administered in the absence of clinical response (O2 saturation) within a further 48 h.
Each patient will be evaluated according to the schedule of assessment illustrated in Table 2.
Adverse event monitoring
Patients will be continuously monitored for adverse events; these will be communicated to the local Transfusion Service, who in turn will notify them to the national electronic system for hemovigilance (SISTRA https://www.centronazionalesangue.it).
Data collection methods
Data will be pseudo-anonymized and they will be collected in a secured web-based database in REDCap that will be built and maintained on a dedicated server hosted at San Matteo Hospital. Data quality and completeness will be monitored using the REDCap query facility for interaction with the clinical investigators. Access to the database will be granted nominally to the investigator(s), who will access via username and password (to be renewed every 2 months). No identification data will be recorded in the database. It will be the responsibility of the principal investigator to maintain an updated list of the patient identification data enrolled in the study together with their enrolment number. Regular back up of the database information will be performed automatically.
Sample size
Considering the hospitalization and mortality data retrieved from the Italian National Institute of Health by March, we expect a mortality of about 15% (survival 85%; H0) in patients treated according to the standard of care. We expect the mortality to decrease to 5% (survival 95%; H1) with the proposed hyperimmune plasma infusion. This being a proof-of-concept study, we use a type I error of 10%. According to the one-stage Fleming design, 43 patients will allow a power above 80% to reject H0. If we will observe at least 40 successes, the H0 hypothesis will be rejected.
For an efficacy-controlled randomized clinical trial, we would have needed to enrol 282 patients (141 per group) to show a decrease in mortality from 15 to 5%.
Statistical analysis
All analyses will be performed using the Stata software (version 16, StataCorp, College Station, TX, USA).
The primary endpoint will be examined by computing the proportion of subjects surviving at 7 days together with its 95% exact binomial confidence interval. Survival will also be described by gender, age < / ≥ 70 years, and comorbidities < / ≥ 3.
Secondary endpoints 1–4 will be described with the median (and quartiles); 95% confidence interval will be retrieved using a quantile regression model. Changes over time of viral load and immune response will be assessed with regression models for repeated measures. Normalization transformations may be applied.
Discussion
The COVID-19 PLASMA trial is the first proof-of-concept interventional trial using hyperimmune plasma with a high titre of specific neutralizing antibodies for treating critical patients with COVID-19. Passive immunization for the prevention and treatment of human infectious diseases and its related concept of artificially acquired passive immunity can be traced back to the twentieth century when specific antibodies were sought from the serum of stimulated animals (especially rabbits and horses). Human blood was also identified as a source of antibodies [4, 15, 16]. Experiences from previous coronavirus epidemics indicate that convalescent plasma collected from recovered COVID-19 patients, containing antibodies against SARS-CoV-2, could be a potentially effective therapeutic weapon [14]. The very recent preliminary communication by Shen and colleagues on five critically ill patients with COVID-19 further confirms the potential benefit of this biologic therapy [7]. With this background, considering the dramatic situation and the high lethality rate of COVID-19 in Italy, we planned an interventional single-arm trial to produce hyperimmune plasma for treating critical patients with COVID-19, in part based on previous experiences in viral infections as H1N1 influenza, Ebola and SARS using plasma infusion or immunoglobulins [4, 5, 9, 10, 13]. Also, the choice of the administration of hyperimmune plasma bears a priori a low risk of adverse events and is readily available.
We hypothesized a potential mechanism of action of plasma from convalescent COVID viremia clearance; this has been confirmed in a recent paper by Rojas et al. [17]. They suggested the direct neutralization of the virus and immunomodulation as the possible mechanisms underlying the beneficial effect of convalescent plasma. For this reason, the optimal therapeutic window needed to account for the simultaneous presence of viremia and a patient’s critical condition. A minimum titer of 1:160 was identified with the aim to administer a sufficient amount of neutralizing antibodies for a prompt patient response. This is in keeping with the FDA and European guidelines [18, 19] successively published.
The main limit of this study is its one-arm design with only 43 patients to be enrolled. However, in the absence of previous evidence, larger and/or randomized trials did not appear to be ethically acceptable. The lack of a control arm does not allow to conclude about the efficacy of the treatment, though this was not the aim of this proof-of-concept study. Informal comparison with the national statistics and concurrent series will add to the decision-making process. The results from this study, if encouraging, will allow us to plan further informed large clinical trials.
Code availability
Not applicable.
Data availability
The data related to the article are available upon request.
References
World Health Organization (2020) Clinical management of severe acute respiratory infection when novel coronavirus (nCoV) infection is suspected. https://www.who.int/docs/default-source/coronaviruse/clinical-management-of-novel-cov.pdf. Accessed 20 Feb 2020.
Clark DR, Jonathan EM, JKB (2020) Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet. https://doi.org/10.1016/S0140-6736(20)30317-2(published online Feb 7)
Holshue ML, DeBolt C, Lindquist S et al (2020) First case of 2019 novel coronavirus in the United States. N Engl J Med. https://doi.org/10.1056/NEJMoa2001191(published online Jan 31)
Marano G, Vaglio S, Pupella S et al (2016) Convalescent plasma: new evidence for an old therapeutic tool? Blood Transfus 14(2):152–157. https://doi.org/10.2450/2015.0131-15
Lu CL, Murakowski DK, Bournazos S et al (2016) Enhanced clearance ofHIV-1 infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science 352(6288):1001–1004. https://doi.org/10.1126/science.aaf1279
Schoofs T, Klein F, Braunschweig M et al (2016) HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science 352(6288):997–1001
Shen C, Wang Z, Zhao F et al (2020) Treatment of 5 critically Ill patients with COVID-19 with convalescent plasma. JAMA. https://doi.org/10.1001/jama.2020.4783
Luke TC, Kilbane EM, Jackson JL, Hoffman SL (2006) Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Ann Intern Med 145(8):599–609. https://doi.org/10.7326/0003-4819-145-8-200610170-00139
Hung IF, To KK, Lee C et al (2011) Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza A (H1N1) 2009 virus infection. Clin Infect Dis 52:447–456. https://doi.org/10.1093/cid/ciq106
Hung IFN, To KKW, Lee CK et al (2013) Hyperimmune IV immunoglobulin treatment: a multicenter double-blind randomized controlled trial for patients with severe 2009 influenza A(H1N1) infection. Chest 144(2):464–473. https://doi.org/10.1378/chest.12-2907
Mair-Jenkins J, Saavedra-Campos M, Baillie J et al (2014) The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. J Infect Dis 211:80–90. https://doi.org/10.1093/infdis/jiu396
Lai S (2005) Treatment of severe acute respiratory syndrome. Eur J Clin Microbiol Infect Dis 24:583–591. https://doi.org/10.1007/s10096-005-0004-z
Soo YO, Cheng Y, Wong R et al (2004) Retrospective comparison of convalescent plasma with continuing high-dose methylprednisolone treatment in SARS patients. Clin Microbiol Infect 10:676–678. https://doi.org/10.1111/j.1469-0691.2004.00956.x
Chen L, Xiong J, Bao L, Shi Y (2020) Convalescent plasma as a potential therapy for COVID-19. Lancet Infect Dis 20(4):398–400. https://doi.org/10.1016/s1473-3099(20)30141-9
Rajam G, Sampson J, Carlone GM, Ades EW (2010) An augmented passive immune therapy to treat fulminant bacterial infections. Recent Pat Antiinfect Drug Discov 5(2):157–167. https://doi.org/10.2174/157489110791233496
Keller MA, Stiehm ER (2000) Passive immunity in prevention and treatment of infectious diseases. Clin Microbiol Rev 13(4):602–614. https://doi.org/10.1128/cmr.13.4.602
Rojas M, Rodriguez Y, Monsalve DM et al (2020) Convalescent plasma in Covid-19: possible mechanisms of action. Autoimmun Rev. https://doi.org/10.1016/j.autrev.2020.102554
https://www.fda.gov/media/136798/download. Accessed 11 May 2020.
https://ec.europa.eu/health/sites/health/files/blood_tissues_organs/docs/guidance_plasma_covid19_en.pdf. Accessed 11 May 2020.
Acknowledgements
We thank Valeria Scotti, librarian at Fondazione IRCCS Policlinico San Matteo, for her support with the references. We also thank all the donors who after experiencing a difficult time being victims of the COVID-19 disease, are freely giving their convalescent plasma for the benefit of all.
Funding
None.
Author information
Authors and Affiliations
Contributions
Substantial contributions to conception and design: all authors. Drafting the article or revising it critically for important intellectual content: CK CDF CP and MF. Final approval of the version to be published: all authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflict of interest.
Ethical approval
Ethical approval was obtained.
Consent to participate
Form approved by Ethical Comm. (protocol).
Consent for publication
Yes.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Perotti, C., Del Fante, C., Baldanti, F. et al. Plasma from donors recovered from the new Coronavirus 2019 as therapy for critical patients with COVID-19 (COVID-19 plasma study): a multicentre study protocol. Intern Emerg Med 15, 819–824 (2020). https://doi.org/10.1007/s11739-020-02384-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-020-02384-2