
Abstract
Prior research has identified abnormal platelet procoagulant responses in COVID-19. Coated-platelets, a form of procoagulant platelets, support thrombin formation and are elevated in ischemic stroke patients with increased risk for recurrent infarction. Our goal was to examine changes in coated-platelet levels over the course of COVID-19 infection and determine their association with disease severity, thrombosis, and death. Coated-platelet levels were assayed after admission and repeated weekly in COVID-19 patients, and in COVID-19 negative controls. Receiver operator characteristic (ROC) analysis was used to calculate area under the curve (AUC) values for a model including baseline coated-platelets to predict death. Kaplan–Meier and Cox proportional hazards analysis was used to predict risk for death at 90 days. We enrolled 33 patients (22 with moderate and 11 with severe infection) and 20 controls. Baseline coated-platelet levels were lower among moderate (mean ± SD; 21.3 ± 9.8%) and severe COVID-19 patients (28.5 ± 11.9%) compared to controls (38.1 ± 10.4%, p < 0.0001). Coated-platelet levels increased during follow-up in COVID-19 patients by 7% (relative) per day from symptom onset (95% CI 2–12%, p = 0.007). A cut-off of 33.9% for coated-platelet levels yielded 80% sensitivity and 96% specificity for death at 90 days, with resulting AUC of 0.880 (95% CI 0.680–1.0, p = 0.0002). The adjusted hazard ratio for death in patients with coated-platelet levels > 33.9% was 40.99 when compared to those with levels ≤ 33.9% (p < 0.0001). Platelet procoagulant potential is transiently decreased in most patients during COVID-19; however, increased baseline platelet procoagulant levels predict death. Defining the mechanisms involved and potential links with aging may yield novel treatment targets.
Similar content being viewed by others


Introduction
SARS-CoV-2 infection and its associated clinical disease, COVID-19, are associated with an aggressive hypercoagulable state, resulting in high rates of deep vein thrombosis and pulmonary embolism despite the use of standard anticoagulant prophylaxis [1, 2]. While severe forms of COVID-19 tend to disproportionately affect the elderly [3], elevated rates of ischemic stroke, myocardial infarction, and peripheral arterial thromboembolism have also been reported, even in younger patients without vascular risk factors [4,5,6,7,8,9].
Growing evidence supports the role of dysfunctional platelet reactivity in the hypercoagulability associated with COVID-19. Altered platelet activation and aggregation due to high levels of inflammatory mediators and immune complexes have been reported [10,11,12]. Autopsy studies have also revealed widespread presence of megakaryocytes and platelet-rich thrombi within small vessels of the hearts, kidneys, and lungs of patients who died of COVID-19 [13,14,15]. Platelets, known to interact with leukocytes and endothelial cells, are visualized within neutrophil extracellular traps (NETs) in COVID-19 lung vessels, implicating platelets as co-mediators of lung damage in acute respiratory distress syndrome (ARDS) [16]. Taken together, these findings support a role for abnormal platelet responses contributing to severe pathology in COVID-19.
Coated-platelets, a subset of platelets produced after co-activation with collagen and thrombin, display enhanced retention of procoagulant proteins on their surface and support thrombin generation [17, 18]. Main characteristics of these activated platelets include surface expression of phosphatidylserine, mitochondrial depolarization, and increased retention of procoagulant proteins, such as fibrinogen, thrombospondin, factor V, fibronectin, and von Willebrand factor [17,18,19,20].
In healthy controls, approximately 30% of circulating platelets express the coated-platelet phenotype upon dual agonist stimulation ex vivo, although the range of individual coated-platelet levels produced is broad and stable over time [21]. Higher levels of coated-platelets are associated with increased risk of recurrent stroke both in large-artery cerebrovascular disease and small-vessel (lacunar stroke) [22, 23] and lower levels are associated with bleeding risk [24,25,26,27,28].
Coated-platelet potential is modified by inflammation, with dramatic increases noted after anti-viral immunization in experimental animals [18]. Similar associations in human populations include positive linear correlations between C-reactive protein and coated-platelet levels among dialysis patients [29], and associations between IL-17A and coated-platelet levels in patients with cerebrovascular disease [30]. Together, these findings suggest that the coated-platelet procoagulant phenotype participates in immunothrombosis, and may play a role in the hypercoagulability in SARS-CoV-2 infection. We now characterize changes in coated-platelet potential over the course of SARS-CoV-2 infection and determine their association with the presence and severity of SARS-CoV-2 infection and with the risk of thrombotic events and death.
Methods and patients
This prospective cohort study was performed at the Veterans Administration Medical Center (VAMC) in Oklahoma City, and was approved by the Institutional Review Board of the University of Oklahoma Health Sciences Center. In accordance with the ethical standards laid down in the Declaration of Helsinki, all subjects or their legally authorized representative provided signed informed consent if the patient was unable to provide signed consent.
Hospitalized subjects with COVID-19 as well as age- and gender-matched hospitalized control subjects without COVID-19 were enrolled. For analysis, COVID-19 subjects were further classified a priori as severe if they were admitted to the intensive care unit, intubated, or died; otherwise, they were classified as moderate COVID-19.
Inclusion criteria for COVID-19 patients consisted of the following: (1) PCR-documented SARS-CoV-2 infection, (2) ability to provide informed consent (from patient or legally authorized representative), and (3) no contraindication for blood draw as judged by the primary treating medical team. Inclusion criteria for controls consisted of the following: (1) PCR-documented negative for SARS-CoV-2 infection and (2) ability to provide individual-signed informed consent.
Blood samples were obtained at the time of enrollment and then repeated weekly during inpatient stay and subsequent outpatient follow-up, if applicable. Coated-platelets were assayed as previously reported [23, 31]. Briefly, platelet-rich plasma was activated simultaneously with thrombin and convulxin. The percentage of platelets with bound biotin-fibrinogen is reported as the coated-platelet level. Laboratory staff members performing the coated-platelet assay were blinded to disease status (COVID-19 positive or negative) and any clinical outcomes.
Outcomes included the following: (1) ST elevation MI, (2) non-ST elevation MI (NSTEMI), (3) myocardial injury, (4) transient ischemic attack (TIA)/stroke, (5) pulmonary embolism (PE), (6) deep vein thrombosis (DVT), and (7) death. MI was defined as a rise and/or fall in troponin I, with at least one value ≥ 0.04 ng/mL, and any one or more of the following: symptoms of myocardial ischemia, new ischemic electrocardiographic changes or Q waves, imaging evidence of new regional wall motion abnormalities or loss of viable myocardium in a pattern consistent with ischemia, and/or evidence of a coronary thrombus via angiography, intracoronary imaging modalities, or by autopsy. Type I and II were differentiated by the suspected underlying mechanism: primary plaque rupture in type I, a primary pathologic process unrelated to coronary thrombosis that results in myocardial oxygen supply and demand mismatch in type II. Lastly, myocardial injury was defined as a rise and/or fall in troponin I, with at least one value ≥ 0.04 ng/mL, not meeting any of the aforementioned criteria [32]. TIA was defined as any transient neurological deficit without imaging evidence of infarction [33]. Stroke was defined as a new, sudden-onset, persistent focal neurological deficit lasting more than 24 h, confirmed by imaging, that could not be attributed to a mass or hemorrhage [34]. PE and DVT were classified as definite if confirmed by objective imaging, or suspected based on clinical presentation when imaging was not possible. Cause of death for all subjects was determined by review of medical records, including death certificates.
All outcomes were established at time of occurrence by the treating physician, who was not involved in the study, and retrieved by chart review. Final adjudication for each outcome was confirmed by a consulting specialist (C.I.P., S.T., K.R.J, S.M.), who was blinded to coated-platelet levels, and recorded in the medical record. Any disagreements were resolved by a blinded adjudicator (MHK or MR) if required.
Sample size estimates and statistical analysis
Sample size was calculated using MedCalc software, Version 19.7 for Windows (MedCalc Software, Ostend, Belgium). We assumed that 20% of subjects would be classified as high risk and 80% as low risk with an overall rate of events of 60% in the high-risk group and 10% in the low-risk group [1, 2, 5]. Using a two-tailed test, p value of 0.05, and power of 0.8, our estimated sample size was 30 [35].
Baseline characteristics were compared between subjects with moderate and severe COVID-19 and non-infected controls using ANOVA or Chi-square tests (or Fisher’s exact tests) as appropriate. Mean coated-platelet levels were compared across infected and control groups using one-way ANOVA and relationships with other clinical variables were assessed using Spearman correlations. Changes in coated-platelet levels over time were estimated using linear mixed models to analyze the change in coated-platelets levels by time and by outcome groups with adjustment for covariates. Receiver operator characteristic (ROC) analysis was used to calculate area under the curve (AUC) values for a model including coated-platelets to predict severe complications or death. A binary coated-platelet cut-point was then identified using Youdon’s index for normally distributed variables. Freedom from death at 90 days was determined by Kaplan–Meier and Cox proportional hazards analysis. Statistical analyses were performed using SAS software, Version 9.4 of the SAS System for Windows (SAS Institute Inc., Cary, NC, USA), R (version 4.0.3) for Windows [36], and GraphPad Prism version 9.0.2 for Windows (GraphPad Software, Sand Diego, CA, USA, www.graphpad.com).
Results
Thirty-three subjects with PCR-positive SARS-CoV-2 infection (22 with moderate COVID-19 and 11 with severe COVID-19) and 20 hospitalized control subjects were enrolled between September 1, 2020, and February 12, 2021.
Age, gender, and race were similarly distributed between moderate COVID-19, severe COVID-19, and hospitalized control groups (Table 1). Vascular risk factors and pertinent medications were not significantly different among the groups (Table 1). Moderate COVID-19 and severe COVID-19 subjects most often presented with pneumonia (18 (85%) and 8 (66%), respectively) whereas hospitalized controls were admitted with bacterial infections (7), cardiovascular disease (4), orthopedic surgery intervention (4), gastrointestinal disease (3), or diabetic ketoacidosis (1).
Follow-up was shorter for severe COVID-19 and hospitalized controls as compared to moderate COVID-19 subjects (mean ± SD, 19.4 ± 13.7 vs. 96.7 ± 52.9 vs. 134.8 ± 47.0 days, p < 0.0001). Time between hospital admission and blood sampling was longer for hospitalized control subjects as compared to severe COVID-19 and moderate COVID-19 subjects (21.7 ± 29.6 vs. 9.3 ± 12.2 vs. 4.7 ± 4.0 days, p = 0.022). Two (10%) hospitalized controls and 10 (83%) severe COVID-19 subjects required intensive care unit admission during hospitalization. Five severe COVID-19 subjects (42%) and 1 hospitalized control (5%) required mechanical ventilation. Four severe COVID-19 subjects (33%) developed acute respiratory distress syndrome compared to none of the moderate COVID-19 or hospitalized control subjects.
Two pulmonary embolism events (one confirmed and one highly suspected but unable to be confirmed) were noted among the severe COVID-19 and moderate COVID-19 subjects, respectively. A portal vein thrombus and a chronic lower extremity deep vein thrombus (DVT) were identified among the hospitalized control subjects, whereas one upper-extremity DVT was diagnosed among the severe COVID-19 subjects. Four moderate COVID-19 (19%) and 5 severe COVID-19 (42%) subjects developed myocardial injury vs. 4 (20%) hospitalized control subjects. One moderate COVID-19 subject developed a TIA at 96 days after symptom onset and 61 days after discharge. Four hospitalized controls (20%) and 1 severe COVID-19 subject (8%) developed major bleeding events. Four hospitalized controls (20%) and 5 severe COVID-19 subjects (42%) died. Of the 5 fatal COVID-19 cases, 2 resulted from progressive hypoxia that did not respond to maximal treatment efforts, 2 were unexpected deaths and occurred suddenly after initial recovery and hospital discharge, and 1 occurred in a moderately severe elderly patient who elected for hospice care early in the hospital course (see Table 2 for a brief description of subjects who died).
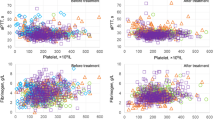
Baseline coated-platelet levels were significantly lower among moderate COVID-19 and severe COVID-19 patients compared to hospitalized control subjects (21.3 ± 9.8 vs. 28.5 ± 11.9 vs. 38.1 ± 10.4%, p < 0.0001, Fig. 1). Among COVID-19 subjects, coated-platelet levels increased during follow-up by an average of 7% (relative) per day from symptom onset (95% CI, 2–12%, p = 0.007) after controlling for age, gender, race, and COVID-19 severity (Fig. 2). Throughout the entire follow-up period, coated-platelet levels were, on average, 13.95% higher at any day from symptom onset among COVID-19 subjects who died compared to those who survived, after adjusting for age, gender, BMI, and race (p = 0.021). No significant difference in coated-platelet levels was detected between moderate and severe COVID-19 subjects (p = 0.308) who survived. Coated-platelet levels were not significantly associated with any pre-specified individual or composite thrombotic outcome.

Distribution of baseline coated-platelet levels in patients with moderate COVID-19, severe COVID-19, and COVID-19 negative controls. Scatter plot showing coated-platelet levels, measured as percentage of the total platelets, in 33 patients with COVID-19, separated into moderate (n = 22) and mild disease (n = 11) categories, and hospitalized COVID-19 negative controls (n = 20). Mean (± standard deviation) coated-platelet levels are lower among moderate COVID-19 and severe COVID-19 patients compared to hospitalized control subjects (21.3 ± 9.8 vs. 28.5 ± 11.9 vs. 38.1 ± 10.4%, p < 0.0001)

Changes in coated-platelet levels during follow-up in COVID-19 patients. Coated-platelet levels in COVID-19 patients increased during the entire period of follow-up by an average of 7% per day from the reported date of symptom onset (95% CI, 2–12%, p = 0.007)
ROC analysis was performed to determine the predictive value of baseline coated-platelet levels among subjects with COVID-19 for death at 90 days after restricting the analysis to 30 subjects whose platelet samples were drawn within 10 days of admission. The resulting AUC was 0.880 (95% CI 0.680–1.0, p = 0.0002), where a cut-point of > 33.9% coated-platelet levels was 80% sensitive and 96% specific for death at 90 days. Cox proportional hazard analysis was performed for predicting risk of death at 90 days using a coated-platelet cut-point of > 33.9% for the high coated-platelet group. This revealed a 40.99 increased hazard rate of death in the high (n = 5) as opposed to the low (n = 25) coated-platelet group (Log Rank χ2 = 25.80, p < 0.0001, Fig. 3).

Probability of survival at 90 days among COVID-19 patients. Five deaths occurred in COVID-19 patients: 4 in patients with coated-platelet levels > 33.9% (red line) and one in a patient with coated-platelet levels ≤ 33.9% (blue line). The curves differ, log-rank chi-square = 25.80, p < 0.0001
Among demographics and clinical factors, we have found that only increased age was also associated with an increased risk for death, with an AUC of 0.836 (95% CI 0.636–1, p = 0.01). There was no significant association between age and coated-platelet levels (p = 0.3).
Discussion
Our results show that coated-platelet levels are significantly lower among hospitalized subjects with moderate to severe COVID-19 compared to hospitalized controls without COVID-19. Moreover, the lower levels noted in COVID-19 subjects increased steadily over time after symptom onset. We have also demonstrated that higher coated-platelet levels within 10 days of hospital admission with COVID-19 are associated with a significant 40.99 increased hazard of death at 90 days.
Several other groups have also demonstrated decreased expression of characteristics associated with the platelet procoagulant phenotype in subjects with COVID-19. Denorme et al. [37] detected significantly lower phosphatidylserine (PS) externalization, reflected by annexin V binding, and decreased mitochondrial depolarization after dual agonist stimulation in COVID-19 subjects compared to healthy control subjects. A statistically significant increase in mitochondrial reactive oxygen species (ROS) was also detected in resting platelets from COVID-19 subjects compared to resting platelets from normal controls in this study [37]. Unlike the platelets from normal controls, levels of ROS failed to increase further after dual agonist stimulation in COVID-19 patients, suggesting that partial activation at baseline may have prevented further changes after dual agonist stimulation.
Others have also demonstrated altered platelet expression of activated αIIb/β3 integrin in subjects with COVID-19. While platelet expression of the active form of this fibrinogen receptor, based on PAC-1 binding, did not differ between COVID-19 patients and controls [38], Taus et al. demonstrated that stimulating platelets from COVID-19 subjects with collagen results in a 60% decrease in PAC-1 binding compared to controls [38]. Manne et al. similarly showed decreased procaspase activating compound-1 (PAC-1) binding after stimulation with 2-methylthio-ADP, thrombin receptor activating peptide, and collagen binding protein at both low and high levels [39]. As suggested by Taus and colleagues [38], it is possible that these findings may be secondary to inactivation of the αIIb/β3 integrin by fibrinogen or fibrin binding in a negative feedback mechanism, similar to findings in influenza and streptococcal pneumonia.
Two additional groups have also independently shown increased PS externalization on resting platelets in COVID-19 patients requiring ICU management compared to non-ICU COVID patients or normal controls [12, 39]. Notably, one of these studies (Althaus et. al.) linked modest increases in PS externalization on resting platelets to higher rates of thrombosis and increased SOFA scores [12]. Increases in PS externalization were not identified by Denorme et al., most likely due to their inclusion of mild to moderate COVID-19 subjects not requiring ICU management [37]. Interestingly, Althaus et al. were able to demonstrate through a series of additional experiments evidence of platelet activation in COVID-19 subjects mediated by platelet FcγRIIA receptor binding by IgG, although antigen specificity of these antibodies remains unknown [12]. Nevertheless, their results also suggest that partial activation of circulating platelets may occur via circulating IgG or IgG immune complexes, which may limit further procoagulant platelet formation upon dual agonist stimulation ex vivo.
Other measures of platelet activation have been identified in subjects with COVID-19. Specifically, increased platelet P-selectin expression on resting platelets and in response to platelet agonists [38,39,40,41], increased platelet-leukocyte aggregates [38, 39], increased binding, and platelet spread on collagen and fibrinogen coated surfaces [39, 40], and increased aggregation with low levels of platelet agonists [38,39,40] have been reported in patients with COVID-19 as compared to healthy donors. It is possible that such activation may predispose platelets in COVID-19 patients for aggregation and adhesion rather than formation of procoagulant platelets, a property that has been demonstrated for coated-platelet formation in other work [42, 43].
In addition to confirming decreased expression of procoagulant platelets in COVID-19, our results extend these previous findings by showing that decreased procoagulant potential in COVID-19 is transient, and that higher levels, rather than lower levels, are highly associated with hazard of death. Because extremes in coated-platelet levels have been associated with occurrence of both thrombotic (elevated levels) and hemorrhagic complications (lower levels) in prior studies [22, 26,27,28, 44], knowledge of an individual’s platelet procoagulant potential during COVID-19 may inform clinical decisions for anticoagulant and antiplatelet prophylaxis and treatment.
While the decrease in platelet procoagulant potential observed is transient (Fig. 2), the length of time required for levels to return to baseline appears to be extend from weeks to months. This may be of interest for COVID-19 patients because prior work has identified significantly lower levels of coated-platelets in stroke patients with MRI evidence of cerebral microbleeds compared to those without such findings [45]. The presence of cerebral microbleeds has been more recently recognized in COVID-19, with rates as high as 30% and unusual locations in patients able to undergo high-resolution imaging [46, 47]. Although mechanisms responsible for these findings are unclear, we hypothesize that decreased levels of procoagulant platelets observed here may be part of the complex mechanisms involved. Given the well-documented link between cerebral microbleeds and presence or progression of vascular cognitive impairment [48], it is very possible that these imaging findings may be linked to the long-term cognitive and behavioral consequences of COVID-19 infection, with specific and significant impact on our aging population.
Additionally, previous work has identified a suppressive effect of antiplatelet therapy with clopidogrel, but not aspirin [31], on coated-platelet levels, suggesting that inhibition of the P2Y12 receptor leads to a sustained decrease in platelet procoagulant potential. Taken together, these findings suggest that longitudinal assessment of platelet procoagulant potential in COVID-19 patients would help identify patients with optimal risk and benefit in ongoing and future clinical trials evaluating antiplatelet therapy [49] in COVID-19.
The potential role for coated-platelet potential in risk stratification in COVID-19 may be further expanded for patients with severe disease, when taking into account recent results reported showing benefits for patients treated with therapeutic enoxaparin [50], and potentially for those receiving treatment with sulodexide, a medication impacting the vascular endothelium, shown to reduce hospital admissions and oxygen requirements when administered in the very early stages of COVID-19 [51].
Surprisingly, we did not detect significant associations between coated-platelet levels and development of DVT, PE, MI, or stroke, although the overall rate of thrombosis in our cohort was low, likely a result of local institutional policies avoiding surveillance vascular imaging in SARS-CoV-2-infected patients. Transthoracic echocardiography and coronary angiography use in SARS-CoV-2-infected patients were also similarly limited, which may lead to an under diagnosis of type I and type II myocardial infarctions. However, of the 5 deaths from COVID-19, 2 occurred in patients with progressive hypoxia, and 2 died abruptly after discharge, which are all characteristics of either in situ or embolic pulmonary thrombosis. Given the strong association in our cohort between elevation of procoagulant platelets and death at 90 days, as well as the clinical picture of the deaths suggestive of fatal thrombotic events, we suspect that higher rates of thrombotic events would have been detected if imaging and autopsy had been more widely available.
We also found that age was a predictor of death in patients with COVID-19. This was not an entirely unexpected finding, since mortality in COVID-19 has been consistently associated with increased age [3]. Since, similar to previous work, no association was present between age and coated-platelet levels, we anticipate that both age and coated-platelet levels may be independent predictors of death on multivariate analysis. Unfortunately, we were limited in the current study by our sample size and were not able to account for multiple variables in our predictive model. Further larger studies, with an adequate representation and range of all age groups, are required to address this possible new issue of an interaction between aging and platelet procoagulant potential.
Additional limitations in our study include lack of autopsy data confirming cause of death and a higher percentage of men as compared to women. A major strength of our study is the serial assessment of platelet procoagulant potential over the hospital admission and outpatient follow-up and longitudinal monitoring of clinical events after discharge.
Nevertheless, the strong association between increased coated-platelet levels and death at 90 days in moderate to severe COVID-19 further supports the link between procoagulant platelet potential and COVID-19.
References
Llitjos JF, Leclerc M, Chochois C, Monsallier JM, Ramakers M, Auvray M, Merouani K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost. 2020;18:1743–6. https://doi.org/10.1111/jth.14869.
Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, Endeman H. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–7. https://doi.org/10.1016/j.thromres.2020.04.013.
Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, the Northwell C-RC, Barnaby DP, Becker LB, Chelico JD, Cohen SL, Cookingham J, Coppa K, Diefenbach MA, Dominello AJ, Duer-Hefele J, Falzon L, Gitlin J, Hajizadeh N, Harvin TG, Hirschwerk DA, Kim EJ, Kozel ZM, Marrast LM, Mogavero JN, Osorio GA, Qiu M, Zanos TP. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323:2052–9. https://doi.org/10.1001/jama.2020.6775.
Madjid M, Safavi-Naeini P, Solomon SD, Vardeny O. Potential effects of coronaviruses on the cardiovascular system: a review. JAMA Cardiol. 2020;5:831–40. https://doi.org/10.1001/jamacardio.2020.1286.
Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, Nigoghossian C, Ageno W, Madjid M, Guo Y, Tang LV, Hu Y, Giri J, Cushman M, Quere I, Dimakakos EP, Gibson CM, Lippi G, Favaloro EJ, Fareed J, Caprini JA, Tafur AJ, Burton JR, Francese DP, Wang EY, Falanga A, McLintock C, Hunt BJ, Spyropoulos AC, Barnes GD, Eikelboom JW, Weinberg I, Schulman S, Carrier M, Piazza G, Beckman JA, Steg PG, Stone GW, Rosenkranz S, Goldhaber SZ, Parikh SA, Monreal M, Krumholz HM, Konstantinides SV, Weitz JI, Lip GYH, Global Covid-19 Thrombosis Collaborative Group EbtINE, the IuaSbtESCWGoPC, Right Ventricular F. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-Up: JACC state-of-the-art review. J Am Coll Cardiol. 2020;75:2950–73. https://doi.org/10.1016/j.jacc.2020.04.031.
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Miao X, Li Y, Hu B. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan China. JAMA Neurol. 2020;77:683–90. https://doi.org/10.1001/jamaneurol.2020.1127.
Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, De Leacy RA, Shigematsu T, Ladner TR, Yaeger KA, Skliut M, Weinberger J, Dangayach NS, Bederson JB, Tuhrim S, Fifi JT. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med. 2020;382:e60. https://doi.org/10.1056/NEJMc2009787.
Bilaloglu S, Aphinyanaphongs Y, Jones S, Iturrate E, Hochman J, Berger JS. Thrombosis in hospitalized patients with COVID-19 in a New York City Health System. JAMA. 2020;324:799–801. https://doi.org/10.1001/jama.2020.13372.
Suh YJ, Hong H, Ohana M, Bompard F, Revel MP, Valle C, Gervaise A, Poissy J, Susen S, Hekimian G, Artifoni M, Periard D, Contou D, Delaloye J, Sanchez B, Fang C, Garzillo G, Robbie H, Yoon SH. Pulmonary embolism and deep vein thrombosis in COVID-19: a systematic review and meta-analysis. Radiology. 2021;298:E70–80. https://doi.org/10.1148/radiol.2020203557.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan China. Lancet. 2020;395:497–506. https://doi.org/10.1016/S0140-6736(20)30183-5.
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76:16–32. https://doi.org/10.1128/MMBR.05015-11.
Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Haberle H, Mehrlander M, Hammer S, Schulze H, Bitzer M, Malek N, Rath D, Bosmuller H, Nieswandt B, Gawaz M, Bakchoul T, Rosenberger P. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood. 2021;137:1061–71. https://doi.org/10.1182/blood.2020008762.
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, Li WW, Li VW, Mentzer SJ, Jonigk D. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383:120–8. https://doi.org/10.1056/NEJMoa2015432.
Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S. COVID-19 autopsies, Oklahoma, USA. Am J Clin Pathol. 2020;153:725–33. https://doi.org/10.1093/ajcp/aqaa062.
Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, Vander Heide RS. Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir Med. 2020;8:681–6. https://doi.org/10.1016/S2213-2600(20)30243-5.
Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, Mostyka M, Baxter-Stoltzfus A, Borczuk AC, Loda M, Cody MJ, Manne BK, Portier I, Harris ES, Petrey AC, Beswick EJ, Caulin AF, Iovino A, Abegglen LM, Weyrich AS, Rondina MT, Egeblad M, Schiffman JD, Yost CC. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136:1169–79. https://doi.org/10.1182/blood.2020007008.
Dale GL, Friese P, Batar P, Hamilton SF, Reed GL, Jackson KW, Clemetson KJ, Alberio L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature. 2002;415:175–9.
Dale GL. Coated-platelets: an emerging component of the procoagulant response. J Thromb Haemostasis. 2005;3:2185–92.
Remenyi G, Szasz R, Friese P, Dale GL. Role of mitochondrial permeability transition pore in coated-platelet formation. Arterioscler Thromb Vasc Biol. 2005;25:467–71.
Dale GL, Remenyi G, Friese P. Quantitation of microparticles released from coated-platelets. J Thromb Haemostasis. 2005;3:2081–8.
Prodan CI, Joseph PM, Vincent AS, Dale GL. Coated-platelet levels are influenced by smoking, aspirin and selective serotonin reuptake inhibitors. J Thromb Haemost. 2007;5:2149–51.
Kirkpatrick AC, Tafur AJ, Vincent AS, Dale GL, Prodan CI. Coated-platelets improve prediction of stroke and TIA in asymptomatic internal carotid artery stenosis. Stroke. 2014;45:2995–3001.
Kirkpatrick AC, Vincent AS, Dale GL, Prodan CI. Increased platelet procoagulant potential predicts recurrent stroke and TIA after lacunar infarction. J Thromb Haemost. 2020;18:660–8. https://doi.org/10.1111/jth.14714.
Brooks MB, Catalfamo JL, Friese P, Dale GL. Scott syndrome dogs have impaired coated-platelet formation and calcein release but normal mitochondrial depolarization. J Thromb Haemost. 2007;5:1972–4.
Saxena K, Pethe K, Dale GL. Coated-platelet levels may explain some variability in clinical phenotypes observed with severe hemophilia. J Thromb Haemost. 2010;8:1140–2.
Daskalakis M, Colucci G, Keller P, Rochat S, Silzle T, Biasiutti FD, Barizzi G, Alberio L. Decreased generation of procoagulant platelets detected by flow cytometric analysis in patients with bleeding diathesis. Cytometry B Clin Cytom. 2014;86:397–409. https://doi.org/10.1002/cytob.21157.
Prodan CI, Stoner JA, Dale GL. Acute hemorrhagic complications are associated with lower coated-platelet levels in non-lacunar brain infarction. J Thromb Haemost. 2015;13:2233–9. https://doi.org/10.1111/jth.13160.
Prodan CI, Stoner JA, Dale GL. Lower coated-platelet levels are associated with increased mortality after spontaneous intracerebral hemorrhage. Stroke. 2015;46:1819–25. https://doi.org/10.1161/STROKEAHA.115.009068.
Valaydon ZS, Lee P, Dale GL, Januszewski AS, Rowley KG, Nandurkar H, Karschimkus C, Best JD, Lyons TJ, Jenkins AJ. Increased coated-platelet levels in chronic haemodialysis patients. Nephrology (Carlton ). 2009;14:148–54.
Kirkpatrick AC, Vincent AS, Guthery L, Nguyen N, Prodan CI, Dale GL. IL-17a Correlates with coated-platelet levels among patients with carotid atherosclerosis. Stroke. 2016;47(suppl 1):ATP 120.
Kirkpatrick AC, Vincent AS, Dale GL, Prodan CI. Clopidogrel use and smoking cessation result in lower coated-platelet levels after stroke. Platelets. 2020;31:236–41. https://doi.org/10.1080/09537104.2019.1609661.
Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD, Executive Group on behalf of the Joint European Society of Cardiology /American College of Cardiology /American Heart Association /World Heart Federation Task Force for the Universal Definition of Myocardial I. Fourth universal definition of myocardial infarction (2018). J Am Coll Cardiol. 2018;72:2231–64. https://doi.org/10.1016/j.jacc.2018.08.1038.
Easton JD, Saver JL, Albers GW, Alberts MJ, Chaturvedi S, Feldmann E, Hatsukami TS, Higashida RT, Johnston SC, Kidwell CS, Lutsep HL, Miller E, Sacco RL. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. The American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke. 2009; 40: 2276-93.
Kernan WN, Ovbiagele B, Black HR, Bravata DM, Chimowitz MI, Ezekowitz MD, Fang MC, Fisher M, Furie KL, Heck DV, Johnston SC, Kasner SE, Kittner SJ, Mitchell PH, Rich MW, Richardson D, Schwamm LH, Wilson JA. Guidelines for the prevention of stroke in patients with stroke and transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:2160–236. https://doi.org/10.1161/STR.0000000000000024.
Machin DC, M.J, Tan SB, Tan SH. Sample size tables for clinical studies. Chinchester: Willey-Blackwell, 2009.
R-CoreTeam. A language and environment for statistical computation. Vienna: R Foundation for Statistical Computing; 2020.
Denorme F, Manne BK, Portier I, Petrey AC, Middleton EA, Kile BT, Rondina MT, Campbell RA. COVID-19 patients exhibit reduced procoagulant platelet responses. J Thromb Haemost. 2020;18:3067–73. https://doi.org/10.1111/jth.15107.
Taus F, Salvagno G, Cane S, Fava C, Mazzaferri F, Carrara E, Petrova V, Barouni RM, Dima F, Dalbeni A, Romano S, Poli G, Benati M, De Nitto S, Mansueto G, Iezzi M, Tacconelli E, Lippi G, Bronte V, Minuz P. Platelets promote thromboinflammation in SARS-CoV-2 pneumonia. Arterioscler Thromb Vasc Biol. 2020;40:2975–89. https://doi.org/10.1161/ATVBAHA.120.315175.
Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, Petrey AC, Tolley ND, Guo L, Cody M, Weyrich AS, Yost CC, Rondina MT, Campbell RA. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136:1317–29. https://doi.org/10.1182/blood.2020007214.
Zaid Y, Puhm F, Allaeys I, Naya A, Oudghiri M, Khalki L, Limami Y, Zaid N, Sadki K, Ben El Haj R, Mahir W, Belayachi L, Belefquih B, Benouda A, Cheikh A, Langlois MA, Cherrah Y, Flamand L, Guessous F, Boilard E. Platelets can associate with sARS-Cov-2 RNA and are hyperactivated in COVID-19. Circ Res. 2020. https://doi.org/10.1161/CIRCRESAHA.120.317703.
Bongiovanni D, Klug M, Lazareva O, Weidlich S, Biasi M, Ursu S, Warth S, Buske C, Lukas M, Spinner CD, Scheidt MV, Condorelli G, Baumbach J, Laugwitz KL, List M, Bernlochner I. SARS-CoV-2 infection is associated with a pro-thrombotic platelet phenotype. Cell Death Dis. 2021;12:50. https://doi.org/10.1038/s41419-020-03333-9.
Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood. 2017;130:2171–9. https://doi.org/10.1182/blood-2017-05-787259.
Aliotta A, Bertaggia Calderara D, Zermatten MG, Alberio L. High-dose epinephrine enhances platelet aggregation at the expense of procoagulant activity. Thromb Haemost. 2021. https://doi.org/10.1055/a-1420-7630.
Brooks MB, Catalfamo JL, Brown HA, Ivanova P, Lovaglio J. A hereditary bleeding disorder of dogs caused by a lack of platelet procoagulant activity. Blood. 2002;99:2434–41.
Prodan CI, Stoner JA, Gordon DL, Dale GL. Cerebral microbleeds in nonlacunar brain infarction are associated with lower coated-platelet levels. J Stroke Cerebrovasc Dis. 2014;23:e325–30. https://doi.org/10.1016/j.jstrokecerebrovasdis.2013.12.016.
Agarwal S, Jain R, Dogra S, Krieger P, Lewis A, Nguyen V, Melmed K, Galetta S. Cerebral microbleeds and leukoencephalopathy in critically ill patients with cOVID-19. Stroke. 2020;51:2649–55. https://doi.org/10.1161/STROKEAHA.120.030940.
Fitsiori A, Pugin D, Thieffry C, Lalive P, Vargas MI. COVID-19 is associated with an unusual pattern of brain microbleeds in critically ill patients. J Neuroimaging. 2020;30:593–7. https://doi.org/10.1111/jon.12755.
Ungvari Z, Tarantini S, Kirkpatrick AC, Csiszar A, Prodan CI. Cerebral microhemorrhages: mechanisms, consequences, and prevention. Am J Physiol Heart Circ Physiol. 2017; 312: https://doi.org/10.1152/ajpheart.00780.2016
Talasaz AH, Sadeghipour P, Kakavand H, Aghakouchakzadeh M, Kordzadeh-Kermani E, Van Tassell BW, Gheymati A, Ariannejad H, Hosseini SH, Jamalkhani S, Sholzberg M, Monreal M, Jimenez D, Piazza G, Parikh SA, Kirtane AJ, Eikelboom JW, Connors JM, Hunt BJ, Konstantinides SV, Cushman M, Weitz JI, Stone GW, Krumholz HM, Lip GYH, Goldhaber SZ, Bikdeli B. Recent randomized trials of antithrombotic therapy for patients with COVID-19: JACC state-of-the-art review. J Am Coll Cardiol. 2021. https://doi.org/10.1016/j.jacc.2021.02.035.
Lemos ACB, Do Espirito Santo DA, Salvetti MC, Gilio RN, Agra LB, Pazin-Filho A, Miranda CH. Therapeutic versus prophylactic anticoagulation for severe COVID-19: a randomized phase II clinical trial (HESACOVID). Thromb Res. 2020;196:359–66. https://doi.org/10.1016/j.thromres.2020.09.026.
Gonzalez-Ochoa AJ, Raffetto JD, Hernandez AG, Zavala N, Gutierrez O, Vargas A, Loustaunau J. Sulodexide in the treatment of patients with early stages of COVID-19: a randomized controlled trial. Thromb Haemost. 2021. https://doi.org/10.1055/a-1414-5216
Acknowledgements
We thank Blair Apple for help with clinical data collection and nursing staff for assistance with consenting and blood draws. We extend our deepest gratitude to all our patients and their families for their willingness to participate in the study.
Funding
The study was funded in part by a grant from the US Department of Veterans Affairs (VA CSR&D COVID19-8900–20).
Author information
Authors and Affiliations
Contributions
Mohamad Khattab, Angelia C. Kirkpatrick, and Calin I. Prodan participated in study design, data collection and classification, patient recruitment, interpretation of results, and manuscript preparation. Andrea S. Vincent and Chao Xu participated in study design, statistical analyses and data presentation, interpretation of results, and manuscript preparation. Eleanor Mathews, Leslie Guthery, and George L. Dale participated in study design, coated-platelet measurements, interpretation of results, and manuscript preparation. Kellie R. Jones, Sharanjeet Thind, and Meheroz Rabadi participated in study design, data classification, and manuscript preparation. All authors gave final approval of the version to be published.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Disclaimer
The funding source had no role in data collection, analysis, or interpretation.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Khattab, M.H., Prodan, C.I., Vincent, A.S. et al. Increased procoagulant platelet levels are predictive of death in COVID-19. GeroScience 43, 2055–2065 (2021). https://doi.org/10.1007/s11357-021-00385-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-021-00385-3