
Abstract
Purpose
This publication reviews the accepted knowledges and the findings still discussed on several features of autoimmune hypophysitis, including the most recently described forms, such as IgG4 and cancer immunotherapy- related hypophysitis.
Methods
The most characteristic findings and the pending controversies were derived from a literature review and previous personal experiences. A single paragraph focused on some atypical examples of the disease presenting under confounding pretences.
Results
Headache, visual field alterations and impaired pituitary secretion are the most frequent clinical findings of the disease. Pituitary biopsy, still considered the gold diagnostic standard, does not always receive consent from the patients. The role of magnetic resonance imaging is limited, as this disease may generate images similar to those of other diseases. The role of antipituitary and antihypothalamus antibodies is still discussed owing to methodological difficulties and also because the findings on the true pituitary antigen(s) are still debated. However, the low sensitivity and specificity of immunofluorescence, one of the more widely employed methods to detect these antibodies, may be improved, considering a predetermined cut-off titre and a particular kind of immunostaining.
Conclusion
Autoimmune hypophysitis is a multifaceted disease, which may certainly be diagnosed by pituitary biopsy. However, the possible different clinical, laboratory and imaging features must be considered by the physician to avoid a misdiagnosis when examining a possibly affected patient. Therapeutic choice has to be made taking into account the clinical conditions and the degree of hypothalamic-pituitary involvement, but also considering that spontaneous remissions can occur.
Similar content being viewed by others


Introduction
Hypophysitis are inflammatory processes of the pituitary gland often involving contiguous structures that are classified as primary and secondary forms [1–5]. The primary forms are characterized by an inflammatory process confined to pituitary gland without a well identifiable etiological agent, while the secondary forms are pituitary inflammatory processes triggered by definite etiologic infective or pharmacological agents or by pituitary involvement of more generalized known systemic diseases (Table 1). On the basis of histopathological findings, primary hypophysitis is divided into five types: lymphocytic hypophysitis (LYH), granulomatous hypophysitis, xanthomatous hypophysitis, necrotizing hypophysitis, and IgG4 plasmacytic hypophysitis, which are still considered truly distinct entities by some authors or only different expressions of the same disease by others because of the possible occurrence of mixed forms [6–10]. LYH, which is the most frequent form of primary hypophysitis, is characterized by an extensive infiltration of the pituitary by lymphocytes and plasma cells [11]. Because of the involved structures it is classified as lymphocytic adenohypophysitis when the process involves only the anterior pituitary, lymphocytic infundibulo-neurohypophysitis when involves the posterior lobe and the infundibulum and lymphocytic panhypophysitis when it involves globally the anterior and the posterior lobe and the infundibulum [12]. A new form of primary hypophysitis has recently been described: IgG4 plasmacytic hypophysitis. This is characterized by the massive infiltration of pituitary gland and/or stalk with numerous IgG4-secreting plasma cells [13]. This disease also called IgG4-related hypophysitis is included, together with several other diseases, in the spectrum of IgG4-related disease [14] (Table 1).
Lymphocytic hypophysytis
Autoimmune hypophysitis is still considered rare, but cases with pituitary autoimmunity are increasingly being recognized. An autoimmune pathogenesis for LYH was proposed by Goudie and Pinkerton in 1962. They described the occurrence of LYH in a young woman showing postpartum amenorrhea and hypothyroidism who died 14 months after delivery for acute hypoadrenalism following surgery for appendicectomy. Autoptic findings showed massive lymphoplasmacytic infiltration of both pituitary and thyroid glands and adrenal atrophy [15]. Actually, a first description had been made by Simmonds [16] in 1917 at pituitary autopsy. Subsequently, a case of panhypopituitarism due to pituitary atrophy with diffuse fibrosis and a moderate lymphocytic infiltration with the lymphocytes collected in small foci, was described by Rupp and Paschkis [17]. However, these authors could not classify this disorder as autoimmune because the concept of endocrine autoimmunity was introduced several years later for Hashimoto’s thyroiditis [18, 19]. From the first description, the frequency of case reports of LYH in the literature is increasing with time. The 1 per 9 million per year incidence estimate, derived from the data by Buxton and Robertson [20] may well be an underestimate of the present incidence of the disease. Caturegli et al. [3] reviewed 379 cases until 2005 and 750 cases until 2008 [21], diagnosed on the basis of clinical and histopathological findings, magnetic resonance imaging (MRI) and detection of pituitary antibodies but the number of new cases described are increasing considerably from these years onwards. This has been favoured by the growing general awareness in the medical community of the condition. Moreover, the recent increase in the use of non-invasive pituitary imaging and of methods to investigate the presence of pituitary antibodies in suspected patients, as well as the pathological findings of pituitary specimens from patients undergoing transsphenoidal surgery, have greatly contributed to the increasing diagnosis of LYH. However, even if several (but not many) cases have appeared in the literature over the years [22–24], at present the true prevalence and incidence of LYH are likely to be underestimated.
Epidemiology
The prevalence of all primary hypophysits is approximately 0.2–0.88 % and the annual incidence is about 1 in 9 million but this is probably underestimated [1–5]. In fact, in the database of the population only cases of biopsy-proven or suspected LYH are included but that the disease is often misdiagnosed has to be considered [3], especially in patients in the subclinical phase.
LYH is the most common form of primary hypophysitis, considering that about 71.8 % of these forms are LYH [2, 3]. LYH is more frequent in women, especially in the last semester of pregnancy and in the post-partum period, with a female/male ratio of about 5:1 [21]. However, cases occurring outside pregnancy have been on the increase in the recent years [3–5, 21]. The mean age at diagnosis is usually 35 ± 13 years for women and 45 ± 14 years for men [1–3], but some cases have been described in children [25] and in the elderly [26]. Among the ethnic factors, in the first series of patients the Caucasian to Japanese ratio was about 3:1 [2] but a recent reevaluation by Caturegli et al. [21] lowered this ratio considerably with increased cases of Japaneses affected, even if still inferior to Caucasians in percentage. The most frequently described HLA alleles were in the first series HLA-DR4 e DR5 [2, 21], but recently HLA-DQ8 and DR53 have been found to be commonly present in patients with LYH [27].
Etiopathogenesis and hystopathology
An autoimmune pathogenesis is suggested by several clinical, laboratory and histopathological findings, even if this is still argument of debate [2–5, 21, 28, 29]. Other endocrine diseases, such as Hashimotos’ thyroiditis, Addison’s disease, type 1 diabetes mellitus and Graves’ disease fulfil the criteria to be considered autoimmune organ-specific diseases. In particular, their respective target autoantigens are well-known tissue-specific or cell-specific enzymes or hormones or receptors capable of evoking their respective autoimmune response [30–32]. Autoimmune hypophysitis is classified as an organ-specific disease because it is caused by selective destruction of pituitary hormone-secreting cells by immune cells. In fact, even if the true pituitary antigen(s) responsible for the immune reaction is (are) still debated, circulating pituitary autoantibodies can be detected in affected patients. Moreover, autoimmune hypophysitis fulfills indirect and circumstantial evidence of autoimmunity according to the established criteria to define a disease as autoimmune, especially regards the histopathological findings [18, 19].
Histopathology
Histopathological findings are suggestive of an autoimmune process overlapping those described for other endocrine glands. Specimens are usually derived at autopsy, pituitary surgery or pituitary biopsy, which is still considered the gold diagnostic standard. They show massive pituitary infiltration by lymphocytes (T more than B lymphocytes, mainly of the CD4 class), plasma cells and macrophages, grouped in aggregates surrounding atrophic acini of pituitary cells [33–35]. Lymphocytes are sometimes arranged in lymphoid follicles with a germinal center, whereas areas of reactive fibrosis can be shown in the remaining pituitary tissue [11] (Fig. 1).

Histopathological findings of autoimmune hypophysitis (modified from Ref. [2])
These germinal centres frequently consist of CD20 positive B cells located centrally with peripheral CD3+ T cells. It has been recently suggested that two distinct entities of LYH can be distinguished on the basis of the prevalence of T-regulatory cells or TH17 cells, which are CD4+ T helper cell effectors in many human autoimmune diseases [35]. One of these entities demonstrates an autoimmune process with TH17 dominance and lack of T-regulatory cells; another form appears as a process in which T-regulatory cells control the immune response which may not be “self-targeted” but rather “foreign targeted” (infective agents?).
Immunochemistry also shows numerous mast cells localized in proximity to capillaries, thus favouring their permeability and angiogenesis, which contribute to perpetuating the inflammatory autoimmune process [36].
Other reasons for considering LYH as an autoimmune disease
Natural history
The natural history of LYH is similar to that of other autoimmune diseases, progressing through several stages with cycles of remission and relapse and with good response to immunosuppressive therapy [37, 38]. Possible spontaneous remission or the progression to the established chronic stage is most likely related to the severity of damage of hypothalamic-pituitary cells and the relationship between the cytokines produced at the sites of the inflammatory autoimmune process and the hypothalamic–pituitary–adrenal (HPA) axis. In fact, these cytokines usually stimulate the HPA axis leading to its hyperactivation with hyperproduction of CRH and AVP, both stimulating directly or indirectly adrenal cortisol secretion, which contributes to the limitation or the extinction of the autoimmune process [2]. When the immune aggression causes severe damage to pituitary cells, mostly ACTH-secreting cells in the early stage of LYH, the consequent impaired secretion of cortisol may not be able to interrupt the immune process. This contributes at perpetuating the aggression to pituitary cells with progression to stable pituitary dysfunction. This condition is even more severe when the autoimmune process involves the hypothalamus, damaging CRH- and AVP-secreting cells, and thus preventing the stimulatory effect of cytokines on the HPA with perpetuation of the inflammatory process. In contrast, when pituitary cell damage is transient, because mostly related to pituitary edema, hyperactivation by the local cytokines of the HPA axis with the consequent cortisol hyperproduction, may interrupt the immune process with recovery of pituitary function [2] (Fig. 2).

Possible self-limitation of the autoimmune pituitary process: cytokines produced at the site of autoimmune inflammatory process stimulate CRH and AVP secretion, both stimulating cortisol secretion, through ACTH hyperproduction(CRH and AVP) or also directly(AVP), which can limit the autoimmune process if the pituitary cells are not irreversibly damaged. PV paraventricular nucleus, SO supraoptic nucleus, AVPs AVP storage in post-pituitary, IL interleukin, TNF tumor necrosis factor (from Ref. [2], with permission)
Association with the autoimmune diseases
Patients with LYH often have a family history of autoimmunity and in some of them LYH shows close association with other autoimmune disease or with autoantibodies to other endocrine glands, configuring in some cases an autoimmune polyendocrine syndrome (APS). Association has been described with several endocrine or non-endocrine diseases [1–3] but the most commonly observed association is with autoimmune thyroid diseases [39] among the APS type 3 or with the diseases falling into the APS type 1 (Fig. 3).

Lymphocytic hypophysitis (LYH) in autoimmune polyendocrine syndromes (APS). The bold characters indicate the diseases most frequently falling into the various types of APS
HLA allele expression in LYH
Several autoimmune diseases are correlated with particular human leukocyte antigen (HLA) alleles [40]. HLA has been typed in a small number of patients with LYH but with results so far inconclusive. In previous studies on 17 patients the predominant allele found was the HLA DR4 and less frequently HLA DR5 [21] but the pituitary cells of none of these patients showed HLA class II molecules, which are often expressed in tissues attacked by autoimmune processes [41, 42]. However, as previously specified, a recent study by Heaney et al. [27] investigated the relationship between specific HLA markers and LYH in 15 patients with sporadic LYH and in 4 patients who had developed hypohysitis after treatment with CTLA 4 antibodies for melanoma, comparing the results with those obtained in patients with other pituitary mass and in normal controls [27]. In patients with sporadic LYH they found HLA-DQ8 expressed in 13 (87 %) and DR53 expressed in 12 (80 %) of 15 patients. In contrast none of the four patients with acquired CTL 4 Ab hypophysitis exhibited the HLA-DQ8 and only 1 of 4 (25 %) exhibited the HLA-DR53 marker. Comparing the results with patients with another pituitary mass, odds ratio of a patient with LYH expressing the HLA-DQ8 marker was 23.1-fold higher than a patient with another sellar mass. These results, on the hand support the autoimmune pathogenesis of LYH, but on the other suggest that HLA-DQ8 testing may assist in the diagnosis of patients with atypical LYH [27].
Diagnostic aspects
The diagnosis of autoimmune hypophysitis is very problematic, as this autoimmune disease can present not only with many different faces, but also because its natural history is very variable [1–5]. During the natural history of the disease an endless series of reversible changes in clinical, morphological and functional findings of the disease can be observed. We describe these diseases including the IgG4-related hypophysitis, and the CTL4 hypophysitis (a new drug-related secondary form of hypophysitis) as forms of autoimmune hypophysitis, because their characteristics tend to overlap with those of LYH.
Clinical and hormonal findings
LYH may present as an acute, subacute or chronic condition with a correspondence between clinical aspects and pituitary involvement in the autoimmune process. Figure 4 summarizes the correlations between the stages of pituitary involvement and clinical and hormonal findings in LYH.

Correlation between pituitary status and clinical and hormonal findings in the different stages of lymphocytic hypophysitis
Acute/subacute phase of LYH
In the acute/subacute phase LYH is characterized by pituitary mass-related symptoms and signs frequently accompanied by symptoms of pituitary failure, which are similar to those of a non-functioning pituitary adenoma [34, 43]. In particular, as regards symptoms of mass effect, headache develops frequently and can occur in a dramatic fashion and then other symptoms, such as visual impairment, nausea and vomiting can be also present [2–5]. Usually the symptoms in this phase are evoked by pituitary edema with extrasellar expansion and pituitary infiltration of lymphocytes and plasma cells. Even if the pituitary biopsy is still considered the gold diagnostic standard for LYH when symptoms of mass effect are present, this is an invasive procedure not always consented to by the patients. Thus, in some patients in the acute/subacute phase, a presumptive diagnosis of LYH could be made on the basis of clinical features and of imaging and laboratory findings [44]. Moreover, as regards isolated or multiple pituitary hormone deficiencies, in these stages they can be sometimes misdiagnosed because the patients are clinically asymptomatic. However, in this phase ACTH is the most frequent and earliest pituitary hormone deficiency, usually associated with hyperprolactinemia. In some rare cases, the rapidity and severity of disease progression may cause a severe secondary adrenal insufficiency with consequent sudden death [1–5]. A multi-factorial etiology has been suggested for the hyperprolactinemia, which affects approximately one-third of patients, causing amenorrhea/galattorrhea in women and sexual dysfunction in men [2, 3]. In some cases it can be due to a decrease in the dopamine delivery to the anterior pituitary related to stalk compression by a pituitary supra-sellar inflammatory mass or to an alteration of dopamine receptors. Hyperprolactinemia can be also present in patients without involvement of extrasellar structures. In these cases, the diffuse lymphocyte infiltration of the pituitary gland could determine an escape of PRL into the circulation secondary to the massive cellular destruction. However, PRL could be directly released by the immune cells infiltrating the gland, considering the relationship between the immune system and PRL [45, 46]. Whatever the cause, the high levels of PRL could contribute to perpetuate the immune process in LYH, through the well-known proinflammatory immunogenic effect of this hormone [45, 46].
Chronic phase of LYH
In the established chronic phase, pituitary fibrosis and atrophy are accompanied by mono or pluritropinic deficiencies with secondary impairment of the respective target gland function. ACTH deficiency may thus be temporally followed by LH, FSH, GH, TSH and more rarely PRL deficiency [47], until a possible panhypopituitarism develops [2–5, 27]. The symptoms and signs are usually not correlated with the severity of pituitary enlargement, suggesting that pituitary hormone deficiencies could be due to the early direct attack of the autoimmune process on the pituitary-secreting cells rather than to later pituitary histopathological alterations [5, 48]. In patients with infundibulo-neurohypopysitis central diabetes insipidus (CDI) is usually present but in some patients with LYH, CDI can be sometimes present, even in the absence of radiological findings of lymphocytic-infundibulo-neurohypophysitis, [2–4, 48].
Recently, the Pituitary Working Group of the German Society of Endocrinology conducted a nationwide retrospective cross-sectional cohort study on the clinical and endocrinological features of primary hypophysitis in 66 German patients with results partially in contrast with those in the literature [49]. Headache (50 %) and weight gain (18 %) were the most frequent nonendocrine symptoms. The association of hypophysitis with pregnancy was observed only in 11 % of the female patients. Diabetes insipidus was found in 54 % of the patients at presentation. Hypogonadotropic hypogonadism was the most frequent endocrine failure (62 %), whereas GH deficiency was the less frequent (37 %). Granulomatous hypophysitis was associated with more severe symptoms than LYH [49]. Even if this German study comprises data from several forms of primary hypophysytis and not from only LYH, the discrepancy between the results from a German population and the data in the literature seems to indicate that environmental factors may influence the clinical features of hypophysytis in affected patients.
Magnetic resonance imaging and its role in differential diagnosis
As previously specified, pituitary biopsy is the gold diagnostic standard for LYH. However, this invasive procedure is not always consented to by the patients. Magnetic resonance imaging (MRI) may help in the diagnosis of LYH and to differentiate this disease from other cases of pituitary mass, in particular pituitary tumor [50, 51], even if imaging findings sometimes tend to overlap, limiting a correct differential diagnosis. In patients with LYH, MRI usually reveals a homogeneous pituitary enlargement, symmetrical supra-sellar extension, compression and displacement of the chiasma, and a thickened, but not deviated, stalk. In German patients thickening of the pituitary stalk is the prevailing neuroradiological sign, being found in 86 % of affected patients [49]. After gadolinium, MRI shows a prompt, intense and homogeneous enhancement of the mass with a strip of enhanced tissue along the dura madre, the so-called: dural tail [50–55]. In patients in whom the process involves the neurohypophysis, the ‘bright spot’ is usually lost and subclinical or clinical CDI is usually present. In some cases, an empty sella can represent an unusual feature of LYH at MRI, particularly during the later stages of the disease [56, 57]. A possible final outcome in empty sella on MRI has been described both for LYH and Shehan’s syndrome (SS) [58]. This syndrome, due to pituitary ischemia after a severe postpartum hemorrage, may be characterized by empty sella on pituitary MRI. The finding of PRL deficiency with post-partum lactational failure has lead to considering a possible diagnosis of SS. However, it is not always possible to differentiate this disease from LYH, because the pituitary ischemia occurring in SS with the consequent cellular damage may trigger an autoimmune process at the pituitary level, which contributes to perpetuate the hypopituitarism in the affected women [59]. In patients with pituitary adenoma, MRI commonly reveals an endosellar mass with unilateral sellar floor depression, asymmetrical supra-sellar extension and controlateral deviation of the stalk. After gadolinium, the enhancement of the pituitary mass is slight, delayed and inhomogeneous. Usually, the dural tail is absent and the bright spot is preserved [50–55] (Table 2).
The above very scholarly criteria do not always provide a correct differential diagnosis. In a further contribution to this argument, Gutemberg et al. [54] some years ago proposed a new radiological score to distinguish autoimmune hypophysitis from not-secreting pituitary adenoma before deciding on a more appropriate therapeutic choice.
In particular, the authors analyzed, by multiple logistic regression, eight features that contributed significantly to classifying pituitary masses as pituitary adenoma or hypophysitis: relation to age, pregnancy, pituitary mass volume and symmetry, signal intensity, signal intensity homogeneity after gadolinium, posterior pituitary bright spot presence, stalk size and mucosal swelling. The score ≥1 suggests a diagnosis of adenoma, whereas the score ≤ 0 suggests a diagnosis of autoimmune hypophysitis. Use of this score resulted in a significant improvement in the differential diagnosis of adenoma and autoimmune hypophysitis compared with the results obtained from the assessment of the single parameters [54]. However, despite the use of the score suggested by Gutenberg et al. [54] the diffferential diagnosis by MRI between LYH and other pituitary mass is still problematical. This was also confirmed by an important paper published in 2011, reporting the results of a 10-year experience on a population of 2598 patients who had been submitted to MRI because of the presence of sellar and parasellar masses [60]. Famini et al. showed that only five patients of 2598 had an LYH, all presenting with panhypopituitarism, but three of these five had been initially diagnosed as having a macroadenoma. These results confirm, on the one hand, the rarity of LYH with respect to the other sellar and parasellar masses of other origin, when investigated by MRI, but on the other hand,also the possibility of misdiagnosis also following the criteria suggested by Gutemberg et al. [54].
Role of antipituitary and antihypothalamus antibodies
Searching for antipituitary antibodies (APA) may help to diagnose or at least suspect an LYH, especially in patients who did not consent a pituitary biopsy and in whom pituitary MRI was inconclusive. However, while other organ-specific antibodies are considered good markers of their respective autoimmune endocrine diseases [61–63], the role of APA in autoimmune hypophysitis is still debated because of several methodological problems and uncertainties in the clinical interpretation [64–67]. Moreover, it has to be considered that the true pituitary antigens reacting with these antibodies are still unknown, even if several antigens have been proposed as responsible for the LYH and as target of APA [67–79]. but their pathogenic role is still argued. Recently, a new target autoantigen, the rabphillin-3A, has been identified as responsible for the autoimmune response in several patients with lymphocytic infundibulo-neurohypophysitis, but also in some patients with LYH [79]. Finally, pituitary-specific transcriptor factor 1(PIT-1) has been shown as a pituitary target of cytotoxic T lymphocytes in a novel clinical entity, the anti-PIT-1 antibody syndrome, that presents an acquired combined pituitary dysfunction characterized by a specific defect in GH, prolactin and TSH. Circulating anti-PIT-1 antibodies along with other various autoantibodies have been detected in some patients with multiple endocrine organopathy, meeting the definition of autoimmune polyglandular syndrome [80].
Methods of detection of antipituitary antibodies
The first method used to detect APA was the complement consumption test [81] but after the first report this method was never employed by other laboratories. The most used methods are immunoblotting assay, radioligand binding and indirect immunofluorescence.
The immunoblotting method [76–78] utilizes a homogenate of human autopsy pituitary tissue as a substrate to identify the antigen target of APA. By this method, Crock [73] showed that serum antibodies against a 49 kDa pituitary cytosolic protein were present in 70 % of patients with biopsy-proven LYH and in 55 % of patients with suspected LYH, including patients with isolated ACTH deficiency, patients with hypopituitarism associated with other autoimmune diseases or females with Sheehan’s syndrome [73]. Subsequently, Crock et al. identified the 49 kDa pituitary cytoplasmatic protein as an α-enolase, which is a ubiquitously expressed enzyme and considered the antibodies to this antigen to be a marker of LYH [77, 78]. Interestingly, α-enolase is expressed in both the pituitary and the placenta, thus providing a theoretic basis for the strong association between pituitary autoimmunity and pregnancy [78]. Subsequent studies have suggested that antibodies to α -enolase cannot be considered specific for LYH because they are frequently present not only in patients with LYH but also in some patients with hypopituitarism secondary to pituitary adenomas or to other pituitary diseases [75].
The technique of radioligand binding assays has led to several studies in organ-specific autoimmune diseases. This method was able to detect autoantibodies against pituitary gland specific factor 1a 306 (PGSF1a) in 33 % patients with biopsy-proven LYH and in 20 % patients with isolated ACTH deficiency [72]. PGSF2 autoantibodies were detected in 14 % patients with suspected LYH or LINH. Anti-GH antibodies were present in 25 % patients with LYH and in 6 % patients with other autoimmune diseases. No patients with pituitary adenoma showed reactivity to either PGSF1a or PGSF2, but PGSF1a autoantibodies were found in 77 % patients with rheumatoid arthritis [73, 74]. However, no information is available on pituitary hormone function in these latter patients and it is not possible to know at present whether these results are due to a lack of specificity of this autoantibody marker or to a high diagnostic sensitivity for a subtle pituitary dysfunction.
Immunofluoresce method is one of the more widely employed methods to detect APA. However, the clinical applicability in the routine diagnosis is still debated for several methodological problems. The main problem of this method is the use of pituitary sections obtained from a variety of species and under a variety of conditions as substrate. In particular, APA were detected in cryostat section of the pituitary gland obtained from humans, primates and rodents and processed with fluorescein isothiocyanate conjugated goat anti-human Ig sera. The influence of human or animal pituitary substrate on the sensitivity and specificity of the immunofluorescence method is still discussed. In fact, immunofluorescence has detected APA not only in some patients with suspected or biopsy-proven LYH, but also in patients with non-autoimmune pituitary diseases such as pituitary adenomas or empty sella syndrome [82–87].
Some authors, by comparing the results obtained from the use of either human pituitary gland or animal pituitary substrates affirmed that the results with animal substrates were inconclusive or had a lower sensitivity and specificity than that with human substrates [65, 82, 83, 88].
Conversely, other authors obtained the same or quite more reliable results using animal than human section [89–91].
Finally, reliable results were obtained by using adrenal and hypothalamic tissues from animals as substrates to search with immunofluorescence for antibodies against key adrenal enzymes or AVP-secreting cells in patients with clinical or subclinical Addison’s disease or CDI, respectively. Thus, these antibodies have been considered good specific, sensible and sometimes predictive markers of Addison’s disease and CDI, respectively [61–63].
Taking into account the previous arguments and aiming to improve the specificity and the sensitivity of the immunoflurescence method, we have established that some characteristics have to be satisfied when processing the immunofluorescence results. In particular, we consider reliable results from immunofluorescence APA detection, by using as substrate cryostat sections of pituitary from young baboon, only when the titre was higher than a cut-off value and when it was associated with a particular kind of immunostaining pattern.
Following these criteria, we searched for APA by immunofluorescence in a large cohort of adults and of children with idiopathic GHD and in patients with idiopathic hypogonadotropic hypogonadism [92–95]. The results of these studies suggested that APA, when present at high titres and with an immunostaining pattern involving selectively some but not all pituitary cells, could be considered good markers of autoimmune isolated hypopitutarism. These antibodies have been further characterized by a four-layer double fluorochrome immunofluorescence test in which, in a second step, the same pituitary section is tested consecutively with the patient’s serum, found positive for APA in the first step, and with the animal’s pituitary hormone antiserum. The different color of the anti-Ig conjugate against the human and the animal serum, respectively green(FITC) and red((rhodamine), allows direct visual assessment of whether the patient’serum and the animal antihormone serum stain the same or different pituitary cells. This allows the identification of the kind of pituitary hormone–secreting cells targeted by APA in patients previously found positive for these antibodies [82, 83]. By this method, we could verify that the pituitary-secreting cells targeted by APA-were somatotrophs in patients with GH deficiency [93, 95] and gonadotrophs in patients with hypogonadotropic hypogonadism [94]. It has been shown that patients with APS or with isolated autoimmune endocrine diseases are at risk of more complex autoimmune diseases, which can be unveiled by searching for the respective autoantibodies [96–99]. Concerning this, in recent studies we have evaluated the predictive value of APA and antihypothalamus antibodies (AHA) detected by simple indirect immunofluorescence, for the occurrence of subsequent hypopituitarism. To this purpose we planned to investigate their time- related variations during a longitudinal study in patients with autoimmune polyendocrine syndromes (APS) positive for these antibodies but with normal pituitary function at diagnosis [95–98]. Our results showed that in some cases, the presence of AHA but not APA was associated with selective or combined hypopituitarism, suggesting that an autoimmune process involving the hypothalamus but not the pituitary, could be responsible for secondary pituitary dysfunction in these patients [98]. On the other hand, the finding of APA at high titre and with immunostaining involving some but not all pituitary cells, were able to predict the occurrence of subsequent hypopituitarism in a follow-up of 5 years in patients with APS without pituitary dysfunction at the diagnosis (97). Moreover, the characterization of pituitary cells targeted by APA in patients with isolated autoimmune diseases, positive for these antibodies at the start but without pituitary dysfunction, may help to foresee the kind of subsequent hypopituitarism [99]. In fact, the detection of APA by double immunofluorescence, selectively directed against gonadotrophs, corticotrophs, thyrotrophs or somatotrophs at the start, was followed during the follow-up by a corresponding pituitary hormone deficiency [99].
Recently, to ascertain the responsibility of hypothalamus or pituitary or both on the pituitary hormone deficiencies in patients with autoimmune hypophysitis positive for APA, the Italian Autoimmune Hypophysitis Network Group performed a study on 95 of these patients, searching for antihypothalamus antibodies [98]. Sera positive for AHA, detected by immunofluorescence, were retested by double immunofluorescence to identify the hypothalamic cells targeted by these antibodies. The results showed that these antibodies were directed not only towards arginine-vasopressin-secreting cell (AVPc) but also towards releasing hormone–secreting cells. In particular, the detection of AHA targeting CRH-secreting cells in patients with GH/ACTH deficiency but with APA specifically directed only to GH-secreting cells, suggested that some pluritropinic deficiencies in some patients with LYH may be due to combined autoimmune aggression both at the pituitary and hypothalamic level [98].
As regards the relationship between pituitary autoimmunity and pregnancy, we recently described a 34 year old woman, who presented with lactational failure after delivery, growth hormone and prolactin deficiencies and subsequently severe primary autoimmune hypothyroidism [100]. Immunological study revealed the presence of APA, identified as targeting GH- and prolactin-secreting cells by double immunofluorescence and elevated levels of antithyroglobulin and antiperoxidase antibodies at the subsequent appearance of thyroid dysfunction. This was the first observation of autoimmune hypopituitarism involving growth hormone and prolactin secretions in a patient with lactational failure after delivery, subsequently followed by severe primary autoimmune hypothyroidism, thus falling into an unusual autoimmune polyendocrine syndrome Type 3. A second similar case has been recently published by Iwama et al. [101]. Pregnancy may also favour the transition from a potential/subclinical to a clinical stage of an autoimmune hypothalamic-pituitary disease. Recently, we studied two women, positive for AVPcAbs before pregnancy but without clinical CDI and who became pregnant 5 and 7 months after our first observation. The behavior of post-pituitary function and AVPcAbs (by immunofluorescence) was evaluated at baseline, during pregnancy and 2 years after delivery [102]. AVPcAbs, present at low/middle titres at baseline in both patients, showed a titre increase during pregnancy in one patient and after delivery in the other one, with development of clinically overt CDI. Therapy with 1-deamino-8-d-AVP (DDAVP) caused a prompt clinical remission. Therapy was definitely stopped at the 6th and 7th month post partum period, respectively, when AVPcAbs disappeared, accompanied by stable post-pituitary function recovery, which persisted until the end of the follow-up. These results indicate that the determination of AVPcAb is advisable in patients with autoimmune diseases planning their pregnancy, because it could be considered a good predictive marker of gestational or post-partum autoimmune CDI. The monitoring of AVPcAb titres and post-pituitary function during pregnancy in these patients may allow an early diagnosis of CDI and an early replacement therapy, which could induce the disappearance of these antibodies with consequent complete remission of autoimmune CDI [102]. An interesting question is whether antipituitary antibodies can change their pituitary target during a life span with consequent change of the kind of pituitary hormone deficiency. Concerning this, we conducted a longitudinal study of pituitary function and APA (by immunoflurescence) on 24 children with apparently idiopathic isolated GH deficiency (GHD), treated with replacement rGH therapy from childhood to the transition age [103]. Pituitary function and APA detection were investigated before starting rGH therapy and after stopping of therapy at transition age. Sera of patients positive for APA were processed by double immunofluorescence to identify their pituitary target. At diagnosis, 16 out of 24 patients were APA positive targeting only somatotrophs (Group 1), while eight were APA negative (Group 2). When retested off therapy 12 of 16 patients in group 1 persisted being APA positive, while the remaining four became APA negative with recovery of pituitary function. All patients in Group 2 persisted APA negative but still showing GHD. Of the 12 patients persistently APA positive, eight with confirmed GHD showed APA still targeting somatotrophs, whereas four showed APA targeting gonadotrophs associated with hypogonadotropic hypogonadism (HH). These results suggest that a possible remission of autoimmune GHD, diagnosed in childhood, may occur in patients positive at middle but not at higher titres at diagnosis, after GH replacement therapy. APA may shift their target in transition period, especially from somatotrophs to gonadotrophs. Thus, an early characterization of APA by double immunofluorescence is advisable in APA positive GHD patients showing delayed puberty to allow an early diagnosis and an appropriate therapy, thus preventing the progression toward a clinically overt HH [103]. Finally, searching for APA in patients with idiopathic hyperprolactinemia may help to disclose the possible occurrence of autoimmune hypophysitis. In these cases, the finding of APA at high titres associated with ACTH or GH deficiency, suggests the diagnosis of LYH in some of them [104–106].
In conclusion, in spite of the diffuse use of the immunofluorescence method, the results appeared in the literature so far, are often conflicting, particularly due to the use of different human or animal substrates. However, we think that improvement of specificity and sensitivity of the method may be obtained, excluding the low titres and the confounding immunostaining patterns. This procedure may allow reliable results for diagnosing or at least suspecting pituitary autoimmunity, by also using animal substrates, especially when the results are validated by a second step with the four-layer double immunofluorescence.
When searching for APA could be useful
We think that searching for APA in some particular conditions, may help to avoid an underestimation of autoimmune hypophysitis; thus, we suggest that the possible presence of APA should be investigated [2, 5]:
-
In patients with apparently idiopathic hypopituitarism especially if associated with other autoimmune diseases
-
In patients with hyperprolactinemia without pituitary adenoma at MRI, without hypothyroidism or associated to iatrogenic causes
-
In patients with hypoprolactinemia and post-partum lactation failure
-
In patients with empty sella
-
In patients with previous traumatic brain injury and in those with infectious meningitis
-
In patients treated with monoclonal antibodies for several types of tumor
-
In patients with IgG4-related syndrome
-
When APA are detected at high titre and with a particular immunofluorescence pattern in these patients, a pituitary MRI (if not yet performed) and a complete evaluation of pituitary function should be performed to discover those with pituitary impairment even at subclinical stage
-
In some cases, searching for antihypothalamus antibodies may help to ascertain the occurrence of an autoimmune process involving selectively the hypothalamic cells or associated with pituitary autoimmunity
IgG4-related hypophysitis
A new form of primary hypophysitis strictly correlated to LYH has been recently described: IgG4 plasmacytic hypophysitis. This disease, also called IgG4-related hypophysitis, is included, with several other diseases, in the spectrum of IgG4-related disease (IgG4-RD). This is an increasingly recognized syndrome of unknown etiology, which comprises a collection of disorders that share specific pathologic, serologic, and clinical features [13, 14, 107]. The commonly shared features include tumor-like swelling of the involved organs, a lymphoplasmacytic infiltrate enriched in IgG4-positive plasma cells and a variable degree of fibrosis. Moreover, elevated serum concentrations of IgG4 are usually found. Several organs may be affected by this disorder. The most frequent presentations are IgG4-related pancreatitis and sclerosing sialoadenitis, but other associated entities have been described, such as sclerosing cholangitis, orbital inflammatory pseudotumor, IgG4-related dacryoadenitis, aortitis and periaortitis, IgG4-related lung disease and mesenteritis, Riedel’s thyroiditis, IgG4-related kidney disease and IgG4-related hypophysitis [14, 107, 108]. In particular, IgG4- related hypophysitis initially was diagnosed in a clinical setting in a 66-year-old woman with inflammatory pseudotumor affecting salivary glands, pancreas and retroperitoneum [13], and also by pathological examination of a 77-year-old man with a history of sclerosing pancreatitis and cholangitis [109]. Subsequently, until 2011 a total of 26 cases of biopsy proven or clinically suspected IgG4-related hypophysitis has been reported [110–112], but further cases have been recently described [113]. This form of hypophysitis is characterized by massive infiltration of the pituitary gland and/or stalk with numerous IgG4-secreting plasma cells [13]. This inflammatory pituitary process can be misdiagnosed as it mimics clinical and imaging features of sellar or parasellar tumor. Following the criteria proposed by Leporati et al. [111], IgG4-related hypophysitis is characterized by the following features: IgG4 serum concentration >135 mg/dl; involvement of other organs; infiltration of IgG4+ plasma cells in the involved tissues, favourable response to glucocorticoid treatment. IgG4-related hypophysitis is more commonly present in men of older age. The more frequent findings are: anterior pituitary enlargement with acute ACTH deficiency when the process involves particularly the pituitary gland; and pituitary stalk thickening with loss of bright spot when the process involves extrasellar structures. In this case CDI may be observed in the majority of the affected patients [18]. A particular case of IgG4-related hypophysitis has been recently described by Hattori et al. [112]. The affected patient presented with loss of bright spot and pituitary stalk thickening on brain MRI but without CDI or anterior pituitary dysfunction. Moreover, he showed a high serum IgG4 level but without other coexisting IgG4-related diseases. An infiltration of lymphocytes and plasma-cells of the posterior lobe, frequently showing IgG4 on their surface but no alterations of the anterior lobe observed on biopsy of either anterior or posterior pituitary lobes [112, 114]. The authors suggested that IgG4- related hypophysitis can be often unrecognized especially in the asymptomatic phase. With regards to CDI, frequently observed in this form of hypophysytis, we found in previous studies that some patients with LYH associated with CDI, were positive for AVPcAb without typical signs on MRI of infundibulo-neurohypophysitis, suggesting that a direct autoimmune attack to AVP-secreting cells could be responsible for their CDI [29, 63]. Following these findings and considering that AVPcAb may belong to IgG4, we think that searching for these antibodies in patients with IgG4-related hypophysitis, could be advisable to clarify the role of these antibodies in their CDI.
CTLA-4 blocking antibodies and hypophysiitis
A secondary form of autoimmune drug-related hypophysitis has been recently described and related to the treatment with cytotoxic T-lymphocyte-associated antigen-4 (CTLA4) blocking antibodies [115–123]. A recent review by Faje summarized the clinical presentation, treatment and biological insight of hypophysitis secondary to immunotherapy [124]. This therapy is frequently employed in the treatment of melanoma and other malignancies. Several autoimmune side effects from these agents have been described, termed immune-related adverse events (IRAEs) and can include multiple endocrinopathies. Among these, immune-targeted cancer therapy-related hypophysitis has been recently recognized as an endocrine IRAE [124, 125]. In fact, recent reports observed that patients developed LYH and other autoimmune diseases during the treatment with the CTLA-4- blocking agent ipulimumab, suggesting a possible correlation between CTLA4 and LYH [115–124]. It has been suggested that these blocking antibodies may act by depleting T-regulatory cells [120]. In another study the antitumor and autoimmune effect of these agents resulted from direct activation of CD4+ CD8+ effector cells [123]. In this context, future studies should be planned to investigate whether a microsatellite polymorphism on the CTLA4 gene, as shown for other autoimmune diseases, could be associated with the development of LYH. Clinical features, including pituitary dysfunction, and MRI of this form are similar to those of LYH. In some cases an electrolytic alteration with hyponatremia has also been described [121]. Usually, following cessation of therapy, a normalization of pituitary morphology at follow-up MRI and return to clinical baseline conditions can be observed spontaneously or after early treatment with high-dose of corticosteroids [122]. Predictive factors for onset of hypophysitis in patients treated with this immunotherapy remain at present unknown. It often presents insidiously with subtle symptoms and can have life-threatening complications, especially related to possible acute hypocortisolism [121–124]. Thus, it is imperative that this form is distinguished from pituitary metastases of melanoma or other malignancies and that a close clinical observation of patient’s progress must be made by physicians. In particular, they have to be cognizant of the overall clinical and radiological picture of this form of hypophysitis, when treating patients receiving ipolimumab or similar drugs with the same effects on the pituitary, to avoid possible severe complications [122, 124].
Autoimmune hypophysitis under false pretences
Unusual presentation of LYH or presentation of other diseases mimicking the features of LYH, may delay the diagnosis with possible damage for the affected patients due to untimely or inappropriate therapy. Here we report some particular presentations appearing in the literature, that may be considered as examples to help avoiding misdiagnosis.
-
Several years ago Lidove et al. described a 36-year-old woman with a prominent lymphoid infiltration of lachrymal and salivary glands, associated with pituitary dysfunction: Laboratory and imaging findings allowed the diagnosis of LYH, which showed a dramatic response to steroids with apparent complete recovery. Instead, 4 years later, Graves’ disease developed, which required an appropriate prolonged therapy. In this case the autoimmune process followed a regional tissue distribution, including pituitary, thyroid, lachrymal and salivary glands [126].
-
Usually the MRI in LYH shows pituitary enlargement with symmetrical extrasellar expansion, with homogeneous and hyperintense signals after gadolinium extending sometimes to the basal hypothalamus in a tongue-like fashion, with stalk thickened but not deviated [50–54]. Instead, Perez-Nunez et al. [127] described in 2005 an interesting case of a patient with LYH presenting with MRI of a cystic lesion with ring contrast enhancement. Since this appearance in imaging studies is not unusual, it should be considered among the possible features suggesting this disease in an appropriate clinical context.
-
Huang et al. [128] described in 2005 a 47-year-old man who had suffered prolonged fever for 2 months without clinical evidence of infection. A pituitary insufficiency was diagnosed by appropriate endocrine investigation. Through whole body imaging studies a 22 by 14 mm mass lesion in the sella turcica was identified, which could suggest the diagnosis of LYH because pathological investigation revealed that the mass was infiltrated by T- and B-lymphoid cells. However, in spite of the timely subtotal tumor resection and steroid supplementation, which induced a gradual fever remission, the mass invaded the cavernous sinus and optic chiasma shortly after surgery. Metastatic lesions in the liver, left adrenal gland and retroperitoneal lymph nodes, were discovered 6 months later. In contrast to cells in the pituitary, liver biopsy revealed the liver mass to be exclusively of T cell origin. The authors concluded that this case exemplifies the rarely noted condition of primary lymphoma with concomitant hypophysitis, whose clinical diagnosis was indiscernible until the occurrence of systemic tumor metastasis [128].
-
Leow et al. [129] performed an interesting study, investigating any chronic endocrine sequelae in 61 survivors from severe acute respiratory syndrome (SARS). The patients were analysed for hormonal derangements 3 months following recovery. Twenthy-four out of 61 patients (39.3 %) had evidence of hypocortisolism, associated with subclinical transient subclinical thyrotoxicosis in two and central hypothyroidism in four of them. The hypothalamic–pituitary–adrenal (HPA) axis dysfunction of the majority resolved within 1 year. These results highlight a possible etiologic role of SARS-associated coronavirus in causing a reversible hypophysitis, with the HPA axis more frequently affected, associated with thyoid autoimmune diseases among an APS type 3.
-
More recently, three interesting cases were published by Kanoke el al [130]. All of them presented with hypophysitis spreading outside pituitary tissue over the cavernous sinus, a very rarely described spreading. Diagnosis of LYH was verified in all three cases at pituitary transphenoidal surgery. Two of the three cases fulfilled the histological criteria of IgG4-related hypophysitis although none of them had high serum IgG4 levels. These cases are the examples of an unusual spreading of hypophysitis to nearby organs [130]. Before this paper only five cases of hypophysitis spreading over the cavernous sinus had been published.
Cases of LYH presenting as temperature dysregulation have been recently published [131, 132].
-
The first case, by Jain and Dhanwa [131], presented with documented body temperature oscillations, hoarseness of voice, sexual dysfunction, pan-hypopituitarism, elevated titre of anti-TPO, postural hypotension and typical findings of LYH on MRI. Prednisone therapy along with hormone replacement therapy induced recovery of a normal rhythm of temperature and of all previous alterations within 30 days.
-
The second case described a complex association of post-partum hypothermia, hypoglicemia, hypoadrenalism and hypothyroidism, accompanied by characteristic findings on MRI. Following these results, the authors suggested that practitioners should keep in mind the possibility of LYH in any pregnant women with symptoms of hypoglicemia and hypothermia after delivery [132].
-
The rupture of Rathke’ cleft cyst may cause lymphocytic-infundibulo-neurohypophysitis. Two cases were published in 2016 by Hayashi et al. [133] presenting with visual disturbances, headaches and endocrine insufficiencies, followed by diabetes insipidus. These are two rare cases of lymphocytic-infundibulo-neurohypophysitis confirmed by post-surgery pathological examination, showing a massive lymphocytic infiltration of the cyst wall and the posterior lobe [133].
-
Specific characteristics of possible autoimmune hypophysitis may also be observed in patients in late phase of complete or selective hypopituitarism, as in patients with Sheehan’s syndrome [59, 60, 134], with hypopituitarism occurring some years after a brain injury [125, 135] or after meningo-encephalitis [136] or in children with celiac disease and short stature, who do not show significant increase in stature on gluten-free diet [96]. In these diseases to search for APA by immunofluorescence may help to disclose a pituitary autoimmunity.
Therapeutic options
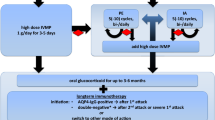
The different expressions of this autoimmune disease require different therapeutic strategies, even because a possible spontaneous remission during the natural history of LYH can occur. For this reason, first, a careful follow-up is advisable in patients without symptomatic extrasellar expansion or important hypoadrenalism [2, 3]. Then, therapeutic options must be different in the acute and chronic phase [4]. In the acute phase, glucocorticoid replacement with stress doses is mandatory in patients with acute adrenal insufficiency. In this phase a surgical option by trans-sphenoidal surgery may be necessary in patients with symptoms and/or signs of severe compression, even if a possible reduction of pituitary mass volume by high doses of methylprednisolone (1 g/day for 3 days), followed by a tapering dose, may be observed [2–4, 129]. Other immunosuppressive drugs, including azathiopirine, methotrexate, cyclosporine A, have also been used successfully, particularly in corticosteroid-resistant cases, but their long-term efficacy still needs to be confirmed [2–4, 21].
Therapeutic strategy in the chronic phase has to be aimed at restoring adequate hormone levels in affected patients with appropriate replacement therapy, recovering secondary hypoadrenalism, hypogonadism, hypothyroidism and GH deficiency, if present, due to LYH per se or as results of neurosurgical therapy [4].
Dopamine agonists (bromocriptine, cabergoline) can lower hyperprolactinemia and improve visual field alterations in some cases but their impact on the course of the disease is still discussed [105]. However, it is well known that high levels of PRL can contribute to perpetuation of the immune process in LYH, through the proinflammatory immunogenic effect of this hormone [45, 46]. This has also recently been demonstrated for hyperprolactinemia related to prolacinoma, which may increase the prevalence of autoimmune diseases, mainly thyroid diseases [137]. Thus, to normalize prolactin levels by dopamine agonists in hypeprolactinemic patients with LYH it should be advisable to prevent possible autoimmune involvement of thyroid or other endocrine glands.
A recent study on the outcome of the main treatment options for LYH has been conducted on 66 German patients, comparing the outcome of the different options. The results showed that glucocorticoid pulse therapy was associated with a high recurrence rate. Moreover, evidence suggested that surgery was not able to prevent recurrence. The authors concluded that, considering the favourable results of observation, conservative management is recommended in LYH unless symptoms are severe or progressive [138].
Recently, rituximab (RTX), a monoclonal antibody that lyzes B cells expressing CD20, usually employed for treatment of several autoimmune diseases, has been employed to treat single cases of steroid-refractory LYH [139, 140]. It was able to induce a remission both in case of isolated LYH [139] and in case of LYH associated with immune thrombocytemia within a polyendocrine syndrome type 4 [140]. Thus, RTX could be considered among the new therapeutic options for the treatment of some type of APS including LYH, provided that these results can be supported by further studies on a large population. Recently, a good response to immunotherapy based on the sequential use of infliximab (a monoclonal antibody that antagonizes tumor necrosis factor alpha) and RTX has been obtained in a 52-year-old woman presenting with LYH occurring with the triad of scleritis, uveitis and optic neuritis, opening new therapeutic horizons for LYH [141].
References
Cheung CC, Ezzat S, Smyth HS, Asa SL (2001) The spectrum and significance of primary hypophysitis. J Clin Endocrinol Metab 86:1048–1053
Bellastella A, Bizzarro A, Coronella C, Bellastella G, Sinisi AA, De Bellis A (2003) Lymphocytic hypophysitis: a rare or underestimated disease? Eur J Endocrinol 149:363–376
Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR (2005) Autoimmune hypophysitis. Endocr Rev 26:599–614
Fukuoka H (2015) Hypophysitis. Endocrinol Metab Clin North Am 44:143–149
De Bellis A, Bellastella G, Colella C, Bizzarro A, Bellastella A, Esposito K (2014) Use of serum pituitary antibodies to improve the diagnosis of hypophysitis. Exp Rev Endocrinol Metab 9:465–476
Scanarini M, d’Ercole AJ, Rotilio A, Kitromilis N, Mingrino S (1989) Giant-cell granulomatous hypophysitis: a distinct clinicopathological entity. J Neurosurg 71:681–686
Vasile M, Marsot-Dupuch K, Kujas M, Brunereau L, Bouchard P, Comoy J, Tubiana JM (1997) Idiopathic granulomatous hypophysitis: clinical and imaging features. Neuroradiology 39:7–11
Deodhare SS, Bilbao JM, Kovacs K, Horvath E, Nomikos P, Buchfelder M, Reschke K, Lehnert H (1999) Xanthomatous hypophysitis: a novel entity of obscure etiology. Endocr Pathol 10:237–241
Cosman F, Post KD, Holub DA, Wardlaw SL (1989) Lymphocytic hypophysitis report of 3 new cases and review of the literature. Medicine 68:240–256
Hassoun P, Anayssi E, Salti I (1985) A case of granulomatous hypophysitis with hypopituitarism and minimal pituitary enlargement. J Neurol Neurosurg Psych 48:949–951
Fehn M, Sommer C, Ludecke DK, Plockinger U, Saeger W (1998) Lymphocytic hypophysitis: light and microscopic findings and correlation to clinical appearance. Endocrine Pathol 9:71–78
Ahmed SR, Aiello DP, Page R, Hopper K, Towfighi J, Santen RJ (1993) Necrotizing infundibulo-hypophysitis: a unique syndrome of diabetes insipidus and hypopituitarism. J Clin Endocrinol Metab 76:1499–1504
van der Vliet HJ, Perenboom RM (2004) Multiple pseudotumors in IgG4- associated multifocal systemic fibrosis. Ann Intern Med 141:896–897
Stone JH, Zen Y, Deshpande V (2012) IgG4-related disease. N Engl J Med 366:539–551
Goudie EB, Pinkerton PH (1962) Anterior hypophysitis and Hashimoto’s disease in a young woman. J Pathol Bacteriol 83:584–585
Simmonds M (1917) Uber das Vorkommen von Riesenzelle in der Hypophyse. Virchows Arch 223:281–290
Rupp JJ, Paschkis KE (1953) Panhypopituitarism with idiopathic hypoparathyroidism. Ann Intern Med 39:1103–1107
Witebsky E, Rose NR, Terplan K, Paine JR, Egan RW (1957) Chronic thyroiditis and autoimmunization. JAMA 164:1439–1447
Rose NR, Bona C (1993) Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol Today 14:426–430
Buxton N, Robertson I (2001) Lymphocytic and granulocytic hypophysitis: a single centre experience. Br J Neurosurg 15:242–246
Caturegli P, Lupi I, Landek-Salgado M, Kimura H, Rose NR (2008) Pituitary autoimmunity: 30 years later. Autoimmun Rev 7:631–637
Rivera JA (2006) Lymphocytic hypophysitis: disease spectrum and approach to diagnosis and therapy. Pituitary 9:35–45
De Bellis A, Ruocco G, Battaglia M, Conte M, Coronella C, Tirelli G, Bellastella A, Pane E, Sinisi AA, Bizzarro A, Bellastella G (2008) Immunological and clinical aspects of lymphocytic hypophysitis. Clin Sci 114:413–421
Falorni A, Minarelli V, Bartoloni E, Alunno A, Gerli R (2014) Diagnosis and classification of autoimmune hypophysitis. Autoimmun Rev 13:412–416
Gellner V, Kurschel S, Scarpatetti M, Mokry M (2008) Lymphocytic hypophysitis in the pediatric population. Childs Nerv Syst 24:785–792
Sellayah R, Gonzales M, Fourlanos S, King J (2015) Lymphocytic hypophysitis in the elderly. J Clin Neurosci 22:1842–1843
Heaney AP, Sumerel B, Rajalingam R, Bergsneider M, Yong WH, Liau LM (2015) HLA markers DQ8 and DR53 are associated with lymphocytic hypophysitis and may aid in differential diagnosis. J Clin Endocrinol Metab 100:4092–4097
De Bellis A, Bizzarro A, Bellastella A (2005) Pituitary antibodies and lymphocytic hypophysitis. Best Pract Res Clin Endocrinol Metab 19:67–84
De Bellis A, Bizzarro A, Bellastella A (2004) Autoimmune central diabetes insipidus. In: Green V, Chrousos G (eds) Immunoendocrinology in health disease. Marcel Dekker, New York, pp 439–459
Palmer JP, Asplin CM, Clemons PL, Tatpati O, Raghu PK, Paquette TL (1983) Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 23:1337–1339
Winqvist O, Karlsson FA, Kämpe O (1992) 21-Hydroxylase, a major autoantigen in idiopathic Addison’s disease. Lancet 339:1559–1562
Weetman AP (2001) Determinants of autoimmune thyroid disease. Nat Immunol 2:769–770
Beressi N, Beressi JP, Cohen R, Modigliani E (1999) Lymphocytic hypophysitis. A review of 145 cases. Ann Intern Med 150:327–341
Ezza S, Josse RG (1997) Autoimmue hypophysitis. Trends Endocrinol Metab 8:74–80
Sautner D, Saeger W, Lüdecke DK, Jansen V, Puchner MJ (1995) Hypophysitis in surgical and autoptical specimens. Acta Neuropathol 90:637–644
Vidal S, Rotondo F, Horvath E, Kovacs K, Scheithauer BW (2002) Immunocytochemical localization of mast cells in lymphocytic hypophysitis. Am J Clin Pathol 117:478–483
De Bellis A, Colao A, Bizzarro A, Di Salle F, Coronella C, Solimeno S, Vetrano A, Pivonello R, Pisano G, Lombardi G, Bellastella A (2002) Longitudinal study of vasopressin-cell antibodies and of hypothalamic-pituitary region on magnetic resonance imaging in patients with autoimmune and idiopathic complete central diabetes insipidus. J Clin Endocrinol Metab 87:3825–3829
De Bellis A, Bizzarro A, Amoresano Paglionico V, Di Martino S, Criscuolo T, Sinisi AA, Lombardi G, Bellastella A (1994) Detection of vasopressin cell antibodies in some patients with autoimmune endocrine diseases without overt diabetes insipidus. Clin Endocrinol 40:173–177
Barbaro D, Loni G (2000) Lymphocytic hypophysitis and autoimmune thyroid disease. J Endocrinol Invest 23:339–340
Ermann J, Fathman CG (2001) Autoimmune diseases: genes, bugs and failed regulation. Nat Immunol 2:759–761
Bottazzo GF, Pujol-Borrell R, Hanafusa T, Feldmann M (1983) Role of aberrant HLA-DR expression and antigen presentation in induction of endocrine autoimmunity. Lancet 2:1115–1119
Todd I, Bottazzo GF (1995) On the issue of inappropriate HLA class II expression on endocrine cells: an answer to a sceptic. J Autoimmun 8:313–322
Khare S, Jagtap VS, Budyal SR, Kasaliwal R, Kakade HR, Bukan A, Sankhe S, Lila AR, Bandgar T, Menon PS, Shah NS (2015) Primary (autoimmune) hypophysitis: a single centre experience. Pituitary 18:16–22
Levy MJ, Jäger HR, Powell M, Matharu MS, Meeran K, Goadsby PJ (2004) Pituitary volume and headache: size is not everything. Arch Neurol 61:721–725
Shelly S, Boaz M, Orbach H (2012) Prolactin and autoimmunity. Autoimmun Rev 11:465–470
De Bellis A, Bizzarro A, Bellastella A (2008) Role of prolactin in autoimmune diseases handbook of systemic autoimmune diseases series. In: Asherson RA, Walker S, Jara LJ (eds) Endocrine manifestations of systemic autoimmune dieseas, vol 9 pp 29–43
Nishiki M, Murakami Y, Ozawa Y, Kato Y (2001) Serum antibodies to human pituitary membrane antigens in patients with autoimmune lymphocytic hypophysitis and infundibuloneurohypophysitis. Clin Endocrinol 54:327–333
Hashimoto K, Takao T, Makino S (1997) Lymphocytic adenohypophysitis and lymphocytic infundibuloneurohypophysitis. Endocrine J 44:1–10
Honegger J, Schlaffer S, Menzel C, Droste M, Werner S, Elbelt U, Strasburger C, Störmann S, Küppers A, Streetz-van der Werf C, Deutschbein T, Stieg M, Rotermund R, Milian M, Petersenn S (2015) Pituitary working group of the German society of endocrinology. Diagnosis of primary hypophysitis in Germany. J Clin Endocrinol Metab 100:3841–3849
Ahmadi J, Meyers GS, Segall HD, Sharma OP, Hinton DR (1995) Lymphocytic adenohypophysitis: contrast-enhanced MR imaging in five cases. Radiology 195:30–34
Chelaifa K, Bouzaidi K, Harzallah F, Menif E, Ben Messaoud M, Turki I, Slim R (2002) Lymphocytic hypophysitis. J Neuroradiol 29:57–60
Nakamura Y, Okada H, Wada Y, Kajiyama K, Koshiyama H (2001) Lymphocytic hypophysitis: its expanding features. J Endocrinol Invest 24:262–267
Sato N, Sze G, Endo K (1998) Hypophysitis: endocrinologic and dynamic MR findings. Am J Neuroradiol 19:439–444
Gutenberg A, Larsen J, Lupi I, Rohde V, Caturegli P (2009) A radiologic score to distinguish autoimmune hypophysitis from nonsecreting pituitary adenoma preoperatively. Am J Neuroradiol 30:1766–1772
Unluhizarci K, Bayrom F, Colak R, Ozturk F, Selcuken A, Durak AC, Kelestimur F (2001) Distinct radiological and clinical appearance of lymphocytic hypophysitis. J Clin Endocrinol Metab 86:1861–1864
Nishiyama S, Takano T, Hidaka Y, Takada K, Iwatani Y, Amino N (1993) A case of postpartum hypopituitarism associated with empty sella: possible relation to postpartum autoimmune hypophysitis. Endocrine J 40:431–438
Klein J, Fehm HL (2005) Unusual presentation of hypophysitis preceding an empty sella in a 75-year-old woman. Neuro Endocrinol Lett 26:757–758
Kelestimur F, Jonsson P, Molvalilai S, Gomez JM, Auemhanmer CJ, Colak R, Koltowska-Haggstrom M, Goth MI (2005) Shehan’s syndrome: baseline characteristics and the effect of 2 years of growth hormone replacement therapy in 91 patients in KIMS_Pfizer International Metabolic Database. Eur J Endocrinol 152:581–587
De Bellis A, Kelestimur F, Sinisi AA, Ruocco G, Tirelli G, Battaglia M, Bellastella G, Conzo G, Tanriverdi F, Unluhizarci K, Bizzarro A, Bellastella A (2008) Anti-hypothalamus and anti-pituitary antibodies may contribute to perpetuate the hypopituitarism in patients with Sheehan’s syndrome. Eur J Endocrinol 158:147–152
Famini P, Maya MM, Melmed S (2011) Pituitary magnetic resonance imaging for sellar and parasellar masses: ten-year experience in 2598 patients. J Clin Endocrinol Metab 96:1633–1641
Pivonello R, De Bellis A, Faggiano A, Di Salle F, Petretta M, Di Somma C, Perrino S, Altucci P, Bizzarro A, Bellastella A, Lombardi G, Colao A (2003) Central diabetes insipidus and autoimmunity: relationship between the occurrence of antibodies to arginine vasopressin-secreting cells and clinical immunological and radiological features in a large cohort of patients with central diabetes insipidus of known and unknown etiology. J Clin Endocrinol Metab 88:1629–1636
De Bellis A, Bizzarro A, Rossi R, Amoresano Paglionico V, Criscuolo T, Lombardi G, Bellastella A (1993) Remission of subclinical adrenocortical failure in subjects with adrenal autoantibodies. J Clin Endocrinol Metab 76:1002–1007
De Bellis A, Colao A, Di Salle F, Muccitelli VI, Iorio S, Perrino S, Pivonello R, Coronella C, Bizzarro A, Lombardi G, Bellastella A (1999) A longitudinal study of vasopressin cell antibodies, posterior pituitary function, and magnetic resonance imaging evaluations in subclinical autoimmune central diabetes insipidus. J Clin Endocrinol Metab 84:3047–3051
Bottazzo GF, Doniach D (1978) Pituitary autoimmunity: a review. J R Soc Med 71:433–436
Gluck M, Scherbaum WA (1990) Substrate specificity for the detection of autoantibodies to anterior pituitary cells in human sera. Horm Metab Res 22:541–545
Maghnie M, Lorini R, Severi F (1994) Antipituitary antibodies in patients with pituitary abnormalities and hormonal deficiency. Clin Endocrinol 40:809–810
Takao T, Nanamiya W, Matsumoto R, Asaba K, Okabayashi T, Hashimoto K (2001) Antipituitary antibodies in patients with lymphocytic hypophysitis. Horm Res 55:288–292
Tzou SC, Lupi I, Landek M, Gutenberg A, Tzou YM, Kimura H, Pinna G, Rose NR, Caturegli P (2008) Autoimmune hypophysitis of SJL mice: clinical insights from a new animal model. Endocrinology 149:3461–3469
Lupi I, Broman KW, Tzou SC, Gutenberg A, Martino E, Caturegli P (2008) Novel autoantigens in autoimmune hypophysitis. Clin Endocrinol 69:269–278
Laway BA, Mir SA (2013) Pregnancy and pituitary disorders: challenges in diagnosis and management. Indian J Endocrinol Metab 17:996–1004
Smith CJ, Bensing S, Burns C, Robinson PJ, Kasperlik-Zaluska AA, Scott RJ, Kämpe O, Crock PA (2012) Identification of TPIT and other novel autoantigens in lymphocytic hypophysitis: immunoscreening of a pituitary cDNA library and development of immunoprecipitation assays. Eur J Endocrinol 166:391–398
Tanaka S, Tatsumi KI, Kimuri M, Takano T, Murakani Y, Takao T, Hashimoto K, Kato Y, Amino N (2002) Detection of autoantibodies against the pituitary-specific protein in patients with lymphocytic hypophysitis. Eur J Endocrinol 147:767–775
Crock P (1998) Cytosolic autoantigens in lymphocytic hypophysitis. J Clin Endocrinol Metab 83:609–618
Tanaka S, Tatsumi KI, Takano T, Murakami Y, Takao T, Yamakita N, Tahara S, Teramoto A, Hashimoto K, Kato Y, Amino N (2003) Anti-alpha-enolase antibodies in pituitary disease. Endocrine J 50:697–702
Bensing S, Fetissov SO, Mulder J, Perheentupa J, Gustafsson J, Husebye ES, Oscarson M, Ekwall O, Crock PA, Hokfelt T, Hulting AL, Kampe O (2007) Pituitary autoantibodies in autoimmune polyendocrine syndrome type 1. Proc Natl Acad Sci USA 104:949–954
Crock P, Salvi M, Miller A, Wall J, Guyda H (1993) Detection of anti-pituitary autoantibodies by immunoblotting. J Immunol Methods 162:31–40
O’Dwyer DT, Smith AI, Matthew ML, Andronicos NM, Ranson M, Robinson PJ, Crock PA (2002) Identification of the 49-kDa autoantigen associated with lymphocytic hypophysitis as α-enolase. J Clin Endocrinol Metab 87:752–757
O’Dwyer DT, Clifton V, Hall A, Smith R, Robinson PJ, Crock PA (2002) Pituitary autoantibodies in lymphocytic hypophysitis target both gamma- and alpha-Enolase—a link with pregnancy? Arch Physiol Biochem 110:94–98
Iwama S, Sugimura Y, Kiyota A, Kato T, Enomoto A, Suzuki H, Iwata N, Takeuchi S, Nakashima K, Takagi H, Izumida H, Ochiai H, Fujisawa H, Suga H, Arima H, Shimoyama Y, Takahashi M, Nishioka H, Ishikawa SE, Shimatsu A, Caturegli P, Oiso Y (2015) Rabphilin-3A as a targeted autoantigen in lymphocytic infundibulo-neurohypophysitis. J Clin Endocrinol Metab 100:946–954
Bando H, Iguchi G, Yamamoto M, Hidaka-Takeno R, Takahashi Y (2015) Anti-PIT-1 antibody syndrome; a novel clinical entity leading to hypopituitarism. Pediatr Endocrinol Rev 12:290–296
Engelberth O, Jezkova Z (1965) Autoantibodies in Sheehan’s syndrome. Lancet 1:1075
Bottazzo GF, Pouplard A, Florin-Christensen A, Doniach D (1975) Autoantibodies to prolactin-secreting cells of human pituitary. Lancet 2:97–101
Bottazzo GF, McIntosh C, Stanford W, Preece M (1980) Growth hormone cell antibodies and partial growth hormone deficiency in a girl with Turner’s syndrome. Clin Endocrinol (Oxford) 12:1–9
Mayfield RK, Levine JH, Gordon L, Powers J, Galbraith RM, Rawe SE (1980) Lymphoid adenohypophysitis presenting as a pituitary tumor. Am J Med 69:619–623
Wild RA, Kepley M (1986) Lymphocytic hypophysitis in a patient with amenorrhea and hyperprolactinemia. A case report. J Reprod Med 31:211–216
Komatsu M, Kondo T, Yamauchi K, Yokokawa N, Ichikawa K, Ishihara M, Aizawa T, Yamada T, Imai Y, Tanaka K (1988) Antipituitary antibodies in patients with the primary empty sella syndrome. J Clin Endocrinol Metab 67:633–638
Mau M, Phillips TM, Ratner RE (1993) Presence of anti-pituitary hormone antibodies in patients with empty sella syndrome and pituitary tumours. Clin Endocrinol 38:495–500
Ricciuti A, De Remigis A, Landek-Salgado MA, De Vincentis L, Guaraldi F, Lupi I, Iwama S, Wand GS, Salvatori R, Caturegli P (2014) Detection of pituitary antibodies by immunofluorescence. Approach and results in patients with pituitary diseases. J Clin Endocrinol Metab 99:1758–1766
Pouplard A (1982) Pituitary autoimmunity. Horm Res 16:289–297
Mauerhoff T, Mirakian R, Bottazzo GF (1987) Autoimmunity and the pituitary. In: Doniach D, Bottazzo GF (eds) Endocrine and other organ-oriented autoimmune disorders. Bailliere’s Clin Immunol Allergy 1:217–235
Cocco C, Brancia C, D’Amato F, Noli B (2014) Pituitary gonadotropins and autoimmunity. Mol Cell Endocrinol 385:97–104
De Bellis A, Bizzarro A, Conte M, Perrino S, Coronella C, Solimeno S, Sinisi AM, Stile LA, Pisano G, Bellastella A (2003) Antipituitary antibodies in adults with apparently idiopathic growth hormone deficiency and in adults with autoimmune endocrine diseases. J Clin Endocrinol Metab 88:650–654
De Bellis A, Salerno M, Conte M, Coronella C, Tirelli G, Battaglia M, Esposito V, Ruocco G, Bellastella G, Bizzarro A, Bellastella A (2006) Antipituitary antibodies recognizing growth hormone (GH)-producing cells in children with idiopathic GH deficiency and in children with idiopathic short stature. J Clin Endocrinol Metab 91:2484–2489
De Bellis A, Sinisi AA, Conte M, Coronella C, Bellastella G, Esposito D, Pasquali D, Ruocco G, Bizzarro A, Bellastella A (2007) Antipituitary antibodies against gonadotropin-secreting cells in adult male patients with apparently idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 92:604–607
De Bellis A, Bizzarro A, Perrino S, Coronella C, Conte M, Pasquali D, Sinisi AA, Betterle C, Bellastella A (2005) Characterization of antipituitary antibodies targeting pituitary hormone-secreting cells in idiopathic growth hormone deficiency and autoimmune endocrine disease. Clin Endocrinol 63:45–49
Delvecchio M, De Bellis A, Francavilla R, Rutigliano V, Predieri B, Indrio F, De Venuto D, Sinisi AA, Bizzarro A, Bellastella A, Iughetti L, Cavallo L (2010) Italian Autoimmune Hypophysitis Network Study Anti-pituitary antibodies in children with newly diagnosed celiac disease: a novel finding contributing to linear-growth impairment. Am J Gastroenterol 105:691–696
Bellastella G, Rotondi M, Pane E, Dello Iacovo A, Pirali B, Della Mora L, Falorni A, Sinisi AA, Bizzarro A, Colao A, Chiovato L, De Bellis A (2010) Predictive role of the immunostaining pattern of immunofluorescence and the titers of antipituitary antibodies at presentation for the occurrence of autoimmune hypopituitarism in patients with autoimmune polyendocrine syndromes over a five-year follow-up. J Clin Endocrinol Metab 95:3750–3757
De Bellis A, Sinisi AA, Pane E, Dello Iacovo A, Bellastella G, Di Scala G, Falorni A, Giavoli C, Gasco V, Giordano R, Ambrosio MR, Colao A, Bizzarro A, Bellastella A (2012) Involvement of hypothalamus autoimmunity in patients with autoimmune hypopituitarism: role of antibodies to hypothalamic cells. Italian Autoimmune Hypophysitis Network Group. J Clin Endocrinol Metab 97:3684–3690
De Bellis A, Dello Iacovo A, Bellastella G, Savoia A, Cozzolino D, Sinisi AA, Bizzarro A, Bellastella A, Giugliano D (2014) Characterization of pituitary cells targeted by antipituitary antibodies in patients with isolated autoimmune diseases without pituitary insufficiency may help to foresee the kind of future hypopituitarsm. Pituitary 17:457–463
De Bellis A, Colella C, Bellastella G, Lucci E, Sinisi AA, Bizzarro A, Holdaway I (2013) Late primary autoimmune hypothyroidism in a patient with postdelivery autoimmune hypopituitarism associated with antibodies to growth hormone and prolactin-secreting cells. Thyroid 23:1037–1041
Iwama S, Welt CK, Romero CJ, Radovick S, Caturegli P (2013) Isolated prolactin deficiency associated with serum autoantibodies against prolactin-secreting cells. J Clin Endocrinol Metab 98:3920–3925
Bellastella G, Bizzarro A, Aitella E, Barrasso M, Cozzolino D, Di Martino S, Esposito K, De Bellis A (2015) Pregnancy may favour the development of severe autoimmune central diabetes insipidus in women with vasopressin cell antibodies: description of two cases. Eur J Endocrinol 172:11–17
De Bellis A, Bellastella G, Maiorino MI, Aitella E, Lucci E, Cozzolino D, Bellastella A, Bizzarro A, Giugliano D, Esposito K (2016) Italian Autoimmune Hypophysitis Network Group. Longitudinal behavior of autoimmune GH deficiency: from childhood to transition age. Eur J Endocrinol 174:381–387
De Bellis A, Bizzarro A, Pivonello R, Lombardi G, Bellastella A (2005) Prolactin and autoimmunity. Pituitary 8:25–30
De Bellis A, Colao A, Pivonello R, Savoia A, Battaglia M, Ruocco G, Tirelli G, Lombardi G, Bellastella A, Bizzarro A (2007) Antipituitary antibodies in idiopathic hyperprolactinemic patients. Ann N Y Acad Sci 1107:129–135
De Bellis A, Colao A, Savoia A, Coronella C, Pasquali D, Conte M, Pivonello R, Bellastella A, Sinisi AA, Bizzarro A, Lombardi G, Bellastella G (2008) Effect of long-term cabergoline therapy on the immunological pattern and pituitary function of patients with idiopathic hyperprolactinaemia positive for antipituitary antibodies. Clin Endocrinol 69:285–291
Hori M, Makita N, Andoh T, Takiyama H, Yajima Y, Sakatani T, Fukumoto S, Iiri T, Fujita T (2010) Long-term clinical course of IgG4-related systemic disease accompanied by hypophysitis. Endocr J 57:485–492
Hirabayashi K, Zamboni G (2012) IgG4-related disease. Pathologica 104:43–55
Wong S, Lam WY, Wong WK, Lee KC (2007) Hypophysitis presented as inflammatory pseudotumor in immunoglobulin G4-related systemic disease. Hum Pathol 38:1720–1723
Aalberse RC, Schuurman J (2002) IgG4 breaking the rules. Immunology 105:9–19
Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P (2011) IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 96:1971–1980
Hattori Y, Tahara S, Ishii Y, Kitamura T, Inomoto C, Osamura RY, Teramoto A, Morita A (2013) A case of IgG4-related hypophysitis without pituitary insufficiency. J Clin Endocrinol Metab 98:1808–1811
Lin W, Lu S, Chen H, Wu Q, Fei Y, Li M, Zhang X, Tian X, Zheng W, Leng X, Xu D, Wang Q, Shen M, Wang L, Li J, Wu D, Zhao L, Wu C, Yang Y, Peng L, Zhou J, Wang Y, Sha Y, Huang X, Jiao Y, Zeng X, Shi Q, Li P, Zhang S, Hu C, Deng C, Li Y, Zhang S, Liu J, Su J, Hou Y, Jiang Y, You X, Zhang H, Yan L, Zhang W, Zhao Y, Zeng X, Zhang F, Lipsky PE (2015) Clinical characteristics of immunoglobulin G4-related disease: a prospective study of 118 Chinese patients. Rheumatology (Oxford) 54:1982–1990
Caturegli P, Iwama S (2013) From Japan with love: another tessera in the hypophysitis mosaic. J Clin Endocrinol Metab 98:1865–1868
Torino F, Barnabei A, Paragliola RM, Marchetti P, Salvatori R, Corsello SM (2013) Endocrine side-effects of anti-cancer drugs: mAbs and pituitary dysfunction: clinical evidence and pathogenic hypotheses. Eur J Endocrinol 23:169–176
Blansfield JA, Beck KE, Tran K, Yang JC, Hughes MS, Kammula US, Royal RE, Topalian SL, Haworth LR, Levy C, Rosenberg SA, Sherry RM (2005) Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother 28:593–598
Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE, Kammula US, Topalian SL, Sherry RM, Kleiner D, Quezado M, Lowy I, Yellin M, Rosenberg SA, Yang JC (2006) Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol 20:2283–2289
Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL, Suri KB, Levy C, Allen T, Mavroukakis S, Lowy I, White DE, Rosenberg SA (2007) Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother 30:825–830
Crowne E, Gleeson H, Benghiat H, Sanghera P, Toogood A (2015) Effect of cancer treatment on hypothalamic-pituitary function. Lancet Diabetes Endocrinol 3:568–576. doi:10.1016/S2213-8587(15)00008-X
Maker AV, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Hughes M, Yellin MJ, Haworth LR, Levy C, Allen T, Mavroukakis SA, Attia P, Rosenberg SA (2006) Intrapatient dose escalation of anti-CTLA-4 antibody in patients with metastatic melanoma. J Immunother 29:455–463
Chodakiewitz Y, Brown S, Boxerman JL, Brody JM, Rogg JM (2014) Ipilimumab treatment associated pituitary hypophysitis: clinical presentation and imaging diagnosis. Clin Neurol Neurosurg 125:125–130
Rodrigues BT, Otty Z, Sangla K, Shenoy VV (2014) Ipilimumab-induced autoimmune hypophysitis: a differential for sellar mass lesions. Endocrinol Diabetes Metab Case Rep 2014:140098
Majchel D, Korytkowski MT (2015) Anticytotoxic T-lymphocyte antigen-4 induced autoimmune hypophysitis: a case report and literature review. Case Rep Endocrinol 2015:570293
Faje A (2016) Immunotherapy and hypophysitis: clinical presentation, treatment, and biologic insights. Pituitary 19:82–92
Melmed S (2016) Pituitary medicine from discovery to patient-focused outcomes. J Clin Endocrinol Metab 101:769–777. doi:10.1210/jc.2015-36532016
Lidove O, Piette JC, Charlotte F, Cassoux N, Correas JM, Papo T (2004) Lymphocytic hypophysitis with lachrymal, salivary and thyroid gland involvement. Eur J Intern Med 15:121–124
Pérez-Núñez A, Miranda P, Arrese I, González P, Ramos A, Lobato RD (2005) Lymphocytic hypophysitis with cystic MRI appearance. Acta Neurochir (Wien) 147:1297–1300
Huang YY, Lin SF, Dunn P, Wai YY, Hsueh C, Tsai JS (2005) Primary pituitary lymphoma presenting as hypophysitis. Endocr J 52:543–549
Leow MK, Kwek DS, Ng AW, Ong KC, Kaw GJ, Lee LS (2005) Hypocortisolism in survivors of severe acute respiratory syndrome (SARS). Clin Endocrinol (Oxf) 63:197–202
Kanoke A, Ogawa Y, Watanabe M, Kumabe T, Tominaga T (2013) Autoimmune hypophysitis presenting with intracranial multi-organ involvement: three case reports and review of the literature. BMC Res Notes 28(6):560
Jain A, Dhanwal DK (2015) A rare case of autoimmune hypophysitis presenting as temperature dysregulation. J Clin Diagn Res 9:OD09-10
Koyama S, Okuno K, Naoi H, Watanabe M, Ozaki K, Shiki Y (2016) Post-partum hypoglycemia and hypothermia as first manifestations of lymphocytic adenohypophysitis: a case report. J Obstet Gynaecol Res 42:467–470
Hayashi Y, Oishi M, Kita D, Watanabe T, Tachibana O, Hamada J (2015) Pure Lymphocytic Infundibuloneurohypophysitis caused by the rupture of Rathke’s Cleft Cyst: report of 2 cases and review of the literature. Turk Neurosurg 25:332–336
Goswami R, Kochupillai N, Crock PA, Jaleel A, Gupta N (2002) Pituitary autoimmunity in patients with Sheehan’s syndrome. J Clin Endocrinol Metab 87:4137–4141
Tanriverdi F, De Bellis A, Bizzarro A, Sinisi AA, Bellastella G, Pane E, Bellastella A, Unluhizarci K, Selcuklu A, Casanueva FF, Kelestimur F (2008) Antipituitary antibodies after traumatic brain injury: is head trauma-induced pituitary dysfunction associated with autoimmunity? Eur J Endocrinol 159:7–13
Tanriverdi F, De Bellis A, Teksahin H, Alp E, Bizzarro A, Sinisi AA, Bellastella G, Paglionico VA, Bellastella A, Unluhizarci K, Doganay M, Kelestimur F (2012) Prospective investigation of pituitary functions in patients with acute infectious meningitis: is acute meningitis induced pituitary dysfunction associated with autoimmunity? Pituitary 15:579–588
Larouche V, Correa JA, Cassidy P, Beauregard C, Garfield N, Rivera J (2016) Prevalence of autoimmune disease in patients with prolactinomas and non-functioning pituitary adenomas. Pituitary 19:202–209
Honegger J, Buchfelder M, Schlaffer S, Droste M, Werner S, Strasburger C, Störmann S, Schopohl J, Kacheva S, Deutschbein T, Stalla G, Flitsch J, Milian M, Petersenn S, Elbelt U (2015) Pituitary Working Group of the German Society of Endocrinology. Treatment of primary hypophysitis in Germany. J Clin Endocrinol Metab 100:3460–3469
Schreckinger M, Francis T, Rajah G, Jagannathan J, Guthikonda M, Mittal S (2012) Novel strategy to treat a case of recurrent lymphocytic hypophysitis using rituximab. J Neurosurg 116:1318–1323
De Bellis A, Colella C, Bellastella G, Savoia A, Guastafierro S, Cozzolino D, Bizzarro A, Bellastella A, Giugliano D (2014) Rituximab-induced remission of autoimmune hypophysitis and primary immune thrombocytopenia in a patient with autoimmune polyendocrine syndrome type 4. Exp Rev Endocrinol Metab. doi:10.1586/17446651.2014.913979
Xu C, Ricciuti A, Caturegli P, Keene CD, Kargi AY (2015) Autoimmune lymphocytic hypophysitis in association with autoimmune eye disease and sequential treatment with infliximab and rituximab. Pituitary 18:441–447
Acknowledgments
The authors wish to thank Dr Trevor G Cooper, Hong Kong SAR, PR China, for his precious suggestions and advices in editing the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Bellastella, G., Maiorino, M.I., Bizzarro, A. et al. Revisitation of autoimmune hypophysitis: knowledge and uncertainties on pathophysiological and clinical aspects. Pituitary 19, 625–642 (2016). https://doi.org/10.1007/s11102-016-0736-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11102-016-0736-z