
Abstract
Aims/hypothesis
Mitochondrial oxidative phosphorylation (OxPhos) is essential for energy production and survival. However, the tissue-specific and systemic metabolic effects of OxPhos function in adipocytes remain incompletely understood.
Methods
We used adipocyte-specific Crif1 (also known as Gadd45gip1) knockout (AdKO) mice with decreased adipocyte OxPhos function. AdKO mice fed a normal chow or high-fat diet were evaluated for glucose homeostasis, weight gain and energy expenditure (EE). RNA sequencing of adipose tissues was used to identify the key mitokines affected in AdKO mice, which included fibroblast growth factor 21 (FGF21) and growth differentiation factor 15 (GDF15). For in vitro analysis, doxycycline was used to pharmacologically decrease OxPhos in 3T3L1 adipocytes. To identify the effects of GDF15 and FGF21 on the metabolic phenotype of AdKO mice, we generated AdKO mice with global Gdf15 knockout (AdGKO) or global Fgf21 knockout (AdFKO).
Results
Under high-fat diet conditions, AdKO mice were resistant to weight gain and exhibited higher EE and improved glucose tolerance. In vitro pharmacological and in vivo genetic inhibition of OxPhos in adipocytes significantly upregulated mitochondrial unfolded protein response-related genes and secretion of mitokines such as GDF15 and FGF21. We evaluated the metabolic phenotypes of AdGKO and AdFKO mice, revealing that GDF15 and FGF21 differentially regulated energy homeostasis in AdKO mice. Both mitokines had beneficial effects on obesity and insulin resistance in the context of decreased adipocyte OxPhos, but only GDF15 regulated EE in AdKO mice.
Conclusions/interpretation
The present study demonstrated that the adipose tissue adaptive mitochondrial stress response affected systemic energy homeostasis via cell-autonomous and non-cell-autonomous pathways. We identified novel roles for adipose OxPhos and adipo-mitokines in the regulation of systemic glucose homeostasis and EE, which facilitated adaptation of an organism to local mitochondrial stress.
Similar content being viewed by others

Introduction
Mitochondria generate the majority of cellular ATP through oxidative phosphorylation (OxPhos), a process in which electrons are transported along five multimeric complexes embedded in the inner mitochondrial membrane to generate a proton gradient for ATP production [1]. Dysregulation of mitochondrial OxPhos is related to metabolic defects such as insulin resistance, impaired beta cell insulin secretion and dysregulation of fatty acid metabolism in mice and humans [2,3,4]. Adipocyte mitochondria play a pivotal role in whole-body metabolism and human diseases [5]. Adipocyte mitochondrial biogenesis and mitochondrial β-oxidation are associated with an improved metabolic phenotype in both rodents and humans [6, 7]. Moreover, proper adipocyte mitochondrial function is required for lipogenesis, lipolysis, and adipokine production and secretion, which regulate systemic energy metabolism [8,9,10]. To maintain and restore proper mitochondrial function, these organelles have evolved a highly conserved mitonuclear communication network, which provides a bidirectional and hormetic response in numerous organisms [11]. However, the adipocyte-specific role of mitochondrial OxPhos in mammals remains unclear.
Recent studies reveal that the OxPhos dysfunction-induced proteotoxic stress activates the mitochondrial unfolded protein response (UPRmt) in vitro and in vivo [12,13,14]. Previously, we demonstrated that deficiency in Crif1 (also known as Gadd45gip1), which encodes mitochondrial large ribosomal subunit protein (CRIF1), leads to abnormal mitochondrial protein homeostasis in mouse embryonic fibroblasts [15]. Deficiency of Crif1 in the brain and in mouse embryonic fibroblasts decreases OxPhos function, enzymatic activity and \( \dot{V}{\mathrm{O}}_2 \) [15]. Furthermore, Crif1-deficient macrophages exhibit reduced basal, ATP-linked and maximal respiration rates compared with wild-type controls [16]. We also found that the \( \dot{V}{\mathrm{O}}_2 \) in Crif1-depleted adipose-derived mesenchymal stem cells was lower than that in controls [17]. Moreover, skeletal muscle-specific Crif1 ablation induces the UPRmt and mitokine production for maintenance of systemic energy homeostasis [18]. Induction of mitochondrial chaperones and proteases as part of the UPRmt is a key mechanism of mitochondrial quality control. In particular, major mitochondrial proteases such as caseinolytic mitochondrial matrix peptidase proteolytic subunit (CLPP) and lon peptidase 1 (LONP1) affect systemic energy metabolism by regulating mitochondrial function and quality control, and by preserving mitochondrial integrity in metabolically active tissues [19, 20]. However, despite high interest in the role of mitochondrial function and quality control in energy metabolism, relatively little is known about the roles of OxPhos function and mitokines in mammalian adipocytes. We, therefore, investigated whether UPRmt and mitokine production caused by lower OxPhos in adipocytes regulates systemic energy metabolism and glucose homeostasis.
Methods
For detailed Methods, please refer to the electronic supplementary material (ESM).
Animal experiments
To generate adipocyte-specific Crif1 knockout mice (AdKO), floxed Crif1 (Crif1f/f) mice were crossed with Adipoq-Cre mice (a kind gift from E. Rosen, Beth Israel Deaconess Medical Center, Boston, MA, USA) on a C57BL/6 background. To generate mitokine double knockout mice, AdKO mice were crossed with global Gdf15−/− or Fgf21−/− on a C57BL/6 background (kindly provided by S-j Lee, Johns Hopkins University School of Medicine, Baltimore, MD, USA; and N. Itoh, Kyoto University Graduate School of Pharmaceutical Sciences, Kyoto, respectively). All animal experiments used male mice and they were fed a normal chow diet (NCD) for 10 weeks or a high-fat diet (HFD, Research Diets, D12492, New Brunswick, NJ, USA) for either 4 or 8 weeks. Mice were started on HFD when they were at 6 weeks of age. GTT and ITT were performed at 8–9 weeks of age. Blood samples were obtained from the 10-week-old (NCD or HFD) or 14-week-old (HFD) mice when they were euthanised. After euthanasia, liver, gastrocnemius, epididymal and inguinal adipose tissue, and brown adipose tissue (BAT) were dissected, weighed and immediately frozen and stored at −80°C or fixed in formalin. The metabolic rate including oxygen consumption (\( \dot{V}{\mathrm{O}}_2 \)), carbon dioxide production (\( \dot{V}{\mathrm{CO}}_2 \)), energy expenditure (EE) and counts of ambulatory physical activity (rearing, activity) in mice were analysed using indirect calorimetry during the day and night periods as previously described [18]. All experimental procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Chungnam National University School of Medicine (CNUH-017-A0048, Daejeon, Korea). See ESM Methods.
GTT and ITT
For evaluating the GTT and ITT, mice were fasted for 16 h and 6 h respectively, and then glucose or insulin was administered by i.p. injection. Serial levels of blood glucose were measured using glucometers. See ESM Methods.
Serum measurements
Blood samples were collected from the hearts of mice under general anaesthesia, and samples were centrifuged at 600 g for 5 min and the supernatant was used for an insulin assay (Alpco Diagnostics, Salem, NH, USA). Growth differentiation factor 15 (GDF15), Fibroblast growth factor 21 (FGF21), adiponectin and leptin were measured using ELISA (R&D Systems, Minneapolis, MN, USA). Levels of serum triacylglycerol, total cholesterol, alanine aminotransferase and aspartate aminotransferase were measured in the wild-type and AdKO mice using a DRI-CHEM 4000i (Fujifilm, Tokyo, Japan).
Histological analysis
Tissue samples including adipose tissue and liver were fixed, processed and stained with H&E. To quantify adipocyte size in adipose tissues, the stained sections were imaged using light microscopy and quantified with Image J software. For immunohistochemistry, tissue sections were incubated with anti-UCP1 antibody (1/200, ab10983, Abcam, Cambridge, UK), and then analysed. See ESM Methods.
Western blot analysis
Proteins including CRIF1, β-actin, α-tubulin, UCP1, OxPhos complex subunits (NADH:ubiquinone oxidoreductase subunit A9 [NDUFA9], NADH:ubiquinone oxidoreductase subunit B8 [NDUFB8], Succinate dehydrogenase complex flavoprotein subunit A [SDHA], ubiquinol–cytochrome c reductase core protein 2 [UQCRC2]) and mitochondrial chaperones and proteases (heat shock 60 kDa protein 1 [HSPD1], DnaJ heat shock protein family (Hsp40) member A3 [DNAJA3], CLPP, LONP1) were detected by immunoblotting with antibodies. Preparation of protein lysates and optimal antibody dilution are indicated in ESM Methods and ESM Table 1, respectively.
Blue native-PAGE
To isolate mitochondria from adipose tissue, the homogenised adipose tissues were prepared to assess the content of OxPhos complexes. Proteins were separated using blue native-PAGE (BN-PAGE) and were transferred to polyvinylidene fluoride membranes, which were incubated overnight with an anti-OxPhos antibody mixture cocktail (Invitrogen, Carlsbad, CA, USA; #45–8099, #45–7999) and analysed using the Western Breeze Chromogenic Western Blot Immunodetection Kit (Invitrogen). See ESM Methods.
Isolation of the stromal vascular fraction and flow cytometry
To analyse macrophage populations in adipose tissue, stromal vascular fractions (SVFs) isolated from adipose tissue were stained with the antibodies listed in ESM Table 2, and then analysed using a FACS Canto II (BD Bioscience). Data were analysed using FlowJo software (FlowJo, Ashland, OR, USA). See ESM Methods.
Adipocyte differentiation
3T3-L1 cells (CL-173) obtained from the ATCC (Manassas, VA, USA) were differentiated with adipogenic cocktail after confirming that the cells were not contaminated with mycoplasma. The cells were treated with doxycycline hydrochloride (Sigma; 10 or 20 μg/ml) for 12 h at 37°C. See ESM Methods.
RNA extraction and quantitative PCR
Total RNA was isolated from adipose tissue. cDNA was synthesised from total RNA and used for measuring the relative expression levels of mRNAs using quantitative PCR. The value was normalised to 18 s rRNA and expressed as a fold change of the value in control extracts. Primer sequences are listed in ESM Table 3. See ESM Methods.
RNA sequencing
Total RNA was extracted from inguinal white adipose tissue (iWAT) using TRIzol reagent and the library was prepared using a TruSeq 3000 4000 SBS Kit, v3 for RNA sequencing analysis. See ESM Methods.
Statistical analysis
All data shown were representative of at least three independent experiments and sample replication of in vivo data was obtained from individual mice. Randomisation and blinding to group assignment and outcome assessment were not carried out in these studies. No results were intentionally removed, data were excluded if they were outside the standard curve range, the sample volume was insufficient, or the sample was lost during the experiment. Statistical analyses were performed using GraphPad Prism 8 (GraphPad, San Diego, CA, USA). Data are expressed as the mean ± SD. All animal data were analysed using a one-way ANOVA (more than two groups) or Student’s two-tailed t test (two groups). A p value <0.05 was considered statistically significant.
Results
AdKO mice demonstrate decreased fat mass but no change in energy expenditure
To determine the impact of adipocyte-specific impairment of mitochondrial OxPhos function on metabolic phenotype, we selectively disrupted Crif1 in adipocytes using the Cre-loxP system. As a result, CRIF1 expression was lower in the epididymal WAT (eWAT), iWAT, and BAT of AdKO mice (Fig. 1a). Consistent with reduced CRIF1 expression, expression of mitochondrial OxPhos complex subunits, including complex I (NDUFB8) and III (UQCRC2), was lower in both eWAT and iWAT of AdKO mice (Fig. 1b and ESM Fig. 1a, b). Moreover, BN-PAGE analysis revealed an apparent decrease in assembly of complexes I, III, and V in AdKO mouse adipose tissue compared with wild-type control mice although we did not calculate the statistical significance in the BN-PAGE analysis due to low sample size (Fig. 1c and ESM Fig. 1c, d).

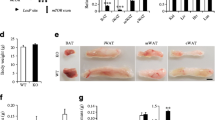
Adipocyte-specific Crif1 knockout is associated with reduced fat mass. (a) Immunoblotting and band density for CRIF1 in eWAT, iWAT, BAT, liver and muscle from NCD-fed Ctrl and AdKO mice at 10 weeks of age (n = 3). (b) Representative blots for OxPhos complex subunits in WAT of NCD-fed Ctrl and AdKO mice at 10 weeks of age. The data were repeated in three independent experiments. (c) Representative blots showing BN-PAGE of the assembled OxPhos complex in WAT from NCD-fed Ctrl and AdKO mice at 10 weeks of age. The arrowhead indicates abnormal sub-complexes. We repeated the runs of the blots three times using the same samples. Quantification of the blots represented in (b) and (c) is shown in ESM Fig. 1. (d) Photographs of whole body, eWAT and iWAT, and graph of body mass gain, of NCD-fed Ctrl and AdKO mice between 6 and 10 weeks of age (n = 4). (e) Organ masses (normalised to body mass) of NCD-fed Ctrl (n = 12) and AdKO (n = 9) mice at 10 weeks of age. (f) Food (NCD) intake (normalised to body mass) over 5 days by Ctrl and AdKO mice at 10 weeks of age (n = 5). (g) Serum leptin concentrations of NCD-fed Ctrl and AdKO mice at 10 weeks of age (n = 5). (h) Representative images of H&E staining of iWAT, eWAT and BAT sections from 10-week-old NCD-fed Ctrl and AdKO mice (n = 4); scale bars, 50 μm. (i, j) Real-time PCR quantification of genes involved in adipogenesis, lipolysis and β-oxidation in (i) iWAT and (j) eWAT from Ctrl and AdKO mice (n = 3). The value was normalised to 18 s rRNA and expressed as a fold change of the value in Ctrl extracts. Data are expressed as the mean ± SD. *p<0.05 vs Ctrl group (and in e vs Ctrl group for the same tissue) by Student’s t test. BW, body weight; CI, CII etc., complex I, II etc.; Ctrl, control
Next, we characterised the metabolic phenotype of AdKO mice on NCD. AdKO mice showed a slight but statistically significant decrease in body mass (22.6 ± 0.5 g) relative to control mice (24.3 ± 1.15 g) at 10 weeks of age (Fig. 1d). Furthermore, after normalising to body mass, eWAT mass was lower in AdKO mice under NCD-fed conditions (Fig. 1e), despite no differences in food intake (Fig. 1f) or serum leptin concentration (Fig. 1g). H&E staining of adipose tissue revealed similarly sized eWAT adipocytes in AdKO and control groups, but AdKO mice had heterogeneous iWAT adipocytes and fewer multilocular adipocytes in the BAT (Fig. 1h).
Levels of eWAT and iWAT transcripts of factors regulating adipocyte differentiation, lipolysis, and β-oxidation were similar in AdKO mice and controls, suggesting that adipocyte differentiation and lipolysis were not compromised by impaired mitochondrial OxPhos (Fig. 1i, j). In addition, liver histology and expression of the liver injury markers alanine aminotransferase and aspartate aminotransferase were similar in AdKO mice and controls (Fig. 2a, b). Serum triacylglycerol and total cholesterol did not differ between the two groups (Fig. 2c, d), but serum adiponectin was significantly lower in AdKO mice (Fig. 2e), which is consistent with prior findings [10].

Adipocyte-specific Crif1 knockout does not affect whole-body EE and glucose homeostasis under normal chow diet-fed conditions. (a) Representative images of H&E staining of liver sections from 10-week-old NCD-fed Ctrl and AdKO mice (n = 4); scale bars, 100 μm (top), 50 μm (enlargements below). (b) Serum alanine aminotransferase and aspartate aminotransferase activities in 10-week-old Ctrl and AdKO mice (n = 4). (c) Serum triacylglycerol and (d) total cholesterol in 10-week-old Ctrl and AdKO mice (n = 4). (e) Serum adiponectin concentration in 10-week-old Ctrl and AdKO mice (n = 5). (f) Counts of total activity and rearing over 3 days in NCD-fed Ctrl and AdKO mice at 10 weeks of age (n = 7). (g) AUC of EE for NCD-fed Ctrl and AdKO mice (n = 4). (h) AUC for \( \dot{V}{\mathrm{O}}_2 \) in 10-week-old Ctrl and AdKO mice fed NCD (n = 4). (i) AUC for \( \dot{V}{\mathrm{CO}}_2 \) in 10-week-old Ctrl and AdKO mice fed NCD (n = 4). (j) Fasting glucose and (k) insulin concentrations (after 16 h of fasting) in NCD-fed Ctrl and AdKO mice (n = 5). (l) GTT and (m) ITT of NCD-fed 10-week-old Ctrl and AdKO mice (n = 5). *p<0.05 vs Ctrl group by Student’s t test. ALT, alanine aminotransferase; AST, aspartate aminotransferase; Ctrl, control
To determine the effect of decreased adipocyte OxPhos on whole-body energy homeostasis, we measured EE using indirect calorimetry. AdKO mice exhibited similar physical activity (Fig. 2f), and had similar total body mass-adjusted EE, \( \dot{V}{\mathrm{O}}_2 \) and \( \dot{V}{\mathrm{CO}}_2 \) (Fig. 2g–i and ESM Fig. 2a–c). These findings implied that decreased body mass in AdKO mice could not be explained by differences in energy intake or EE. Furthermore, there were no significant differences in fasting glucose or serum insulin (Fig. 2j, k), or in glucose or insulin tolerance (as determined by GTT and ITT, respectively), between the two groups (Fig. 2l, m).
Taken together, these findings suggest that lower OxPhos function in adipocytes results in decreased body mass due to reduction in adipose tissue mass, but does not affect systemic EE or glucose homeostasis under NCD-fed conditions. To exclude the possibility of temperature-dependent effects, we performed indirect calorimetry to analyse the EE of control and AdKO mice under thermoneutral conditions. In this system, AdKO and control mice showed no differences in EE, body weight, or tissue weight compared with the results from room temperature experiments (ESM Fig. 3a–c).
AdKO mice are protected against HFD-induced obesity and insulin resistance
To determine the systemic effects of decreased adipocyte OxPhos in the context of metabolic stress, AdKO mice and controls were fed an HFD for 4 weeks. Weight gain was significantly decreased in AdKO mice relative to controls (Fig. 3a). After normalising to body mass, iWAT and eWAT masses were significantly lower, but BAT mass was higher, in AdKO mice (Fig. 3b). H&E staining of adipose tissues revealed that WAT adipocyte size was significantly decreased in HFD-fed AdKO mice relative to that in HFD-fed controls, but BAT adipocyte size was similar (Fig. 3c). However, in AdKO mice there was significantly reduced mRNA expression of Cebpa, Plin1, Lipe and Pnpla2 (which are involved in the regulation of adipogenesis and lipolysis) in iWAT only (ESM Fig. 4a, b). These results suggest that decreased adipocyte OxPhos capacity induces reduction of adipogenesis, consistent with previous data [17], and that decreased lipolysis inhibits ectopic lipid accumulation in non-adipose tissues in the HFD-fed condition. Interestingly, serum concentrations of adiponectin and leptin, as well as circulating triacylglycerol and total cholesterol, were much lower in AdKO mice (ESM Fig. 4c–f). Reduction of fat mass in AdKO mice may have reduced serum leptin levels, which is consistent with a prior report of a positive correlation between obesity or insulin resistance and serum leptin levels in humans [21]. In addition, hepatic fat accumulation was attenuated markedly in AdKO mice (Fig. 3d). Fasting blood glucose was lower in AdKO mice (Fig. 3e), while fasting insulin concentration was similar between groups (Fig. 3f). To determine the effects of impaired adipocyte OxPhos function on systemic glucose metabolism, we measured glucose tolerance, which was improved in AdKO mice vs controls (Fig. 3g). Moreover, ITT revealed that insulin sensitivity was improved in AdKO mice relative to control mice (Fig. 3h). These data suggest that adipocyte-specific impairment of OxPhos improves glucose metabolism and protects against diet-induced obesity and insulin resistance in the context of HFD feeding.

Adipocyte-specific Crif1 knockout protects against obesity and improves systemic metabolism in mice consuming an HFD. (a) Photographs of whole body, eWAT and iWAT and graph of body mass gain in HFD-fed Ctrl and AdKO mice between 6 and 10 weeks of age; 6-week-old mice were fed an HFD for 4 weeks (n = 5). (b) Organ masses (normalised to body mass) for Ctrl and AdKO mice after 4 weeks of HFD feeding (n = 5). (c) Representative images of H&E staining of adipose sections of WAT and BAT from 10-week-old Ctrl and AdKO mice fed an HFD for 4 weeks (n = 5); scale bars, 50 μm. (d) Representative images of H&E staining of liver sections from 10-week-old Ctrl and AdKO mice fed an HFD for 4 weeks (n = 5); scale bars, 100 μm (top), 50 μm (enlargements below). (e) Fasting glucose and (f) insulin concentrations (after 16 h of fasting) in 10-week-old Ctrl and AdKO mice fed an HFD for 4 weeks (n = 4). (g) GTT and (h) ITT in 10-week-old Ctrl and AdKO mice fed an HFD for 4 weeks (n = 4). *p < 0.05 vs Ctrl HFD group (and in b vs Ctrl HFD for the same tissue) by Student’s t test. BW, body weight; Ctrl, control
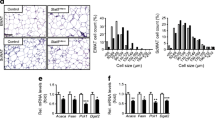
The extent of macrophage infiltration into adipose tissue governs the local and systemic inflammatory responses that contribute to insulin resistance [22]. To compare the macrophage population and its functional status in AdKO mice and controls, we performed FACS analysis on the adipose SVF using the macrophage surface markers CD11c (an M1 marker) and CD206 (an M2 marker). Using this approach, we found that AdKO mice had a larger iWAT macrophage population, and more specifically, larger numbers of M2 macrophages (Fig. 4a–c).

Adipocyte-specific Crif1 knockout increases EE and elevates UCP1 expression in WAT. (a) Representative flow cytometry plots of M1 (CD11c+CD206−) and M2 (CD11c−CD206+) macrophage populations in total WAT SVF immune cells from 10-week-old Ctrl and AdKO mice fed an HFD for 4 weeks (n = 3). (b, c) Macrophage cell number per gram of (b) iWAT and (c) eWAT in 10-week-old Ctrl and AdKO mice fed an HFD for 4 weeks (n = 3). (d) Immunoblotting for UCP1 in WAT and BAT and graphs showing quantification of band density of UCP1 protein in iWAT and eWAT (normalised to β-actin) and BAT (normalised to α-tubulin) from 10-week-old Ctrl (n = 4) and AdKO (n = 5) mice fed an HFD for 4 weeks. (e) AUC for EE in 10-week-old Ctrl (n = 4) and AdKO (n = 3) mice fed an HFD for 4 weeks. (f) AUC for \( \dot{V}{\mathrm{O}}_2 \) in 10-week-old Ctrl and AdKO mice fed an HFD for 4 weeks. (g) AUC for \( \dot{V}{\mathrm{CO}}_2 \) in 10-week-old Ctrl and AdKO mice fed an HFD for 4 weeks. *p<0.05 vs Ctrl HFD group (and in b and e–g vs Ctrl HFD group for the same category) by Student’s t test. Ctrl, control
Next, we measured iWAT and eWAT uncoupling protein 1 (UCP1) expression in AdKO mice and controls fed an HFD for 4 weeks and found that UCP1 levels in AdKO mice were significantly higher in WAT, but significantly lower in BAT, than levels in controls (Fig. 4d). Immunostaining further confirmed that WAT UCP1 levels were increased in AdKO mice (ESM Fig. 5). To determine the systemic effects of these differences in UCP1 expression, we measured EE in AdKO and control mice. Interestingly, AdKO mice had higher EE as well as \( \dot{V}{\mathrm{O}}_2 \) and \( \dot{V}{\mathrm{CO}}_2 \) (adjusted to total body mass) during the dark phase (Fig. 4e–g and ESM Fig. 6a–c) suggesting that increased uncoupling in iWAT and eWAT could contribute to higher EE in AdKO mice.
Genetic or pharmacological inhibition of adipocyte mitochondrial OxPhos function induces the UPRmt in vitro and in vivo
Mitochondrial OxPhos deficits cause proteotoxic stress, which initiates the UPRmt to induce mitochondrial proteostasis, a highly conserved mitoprotective mechanism [13, 23]. Therefore, we measured expression of mitochondrial chaperones and proteases in AdKO mice and controls under both NCD- and HFD-fed conditions. Expression of iWAT and eWAT Lonp1 mRNA increased in NCD-fed AdKO mice (ESM Fig. 7a, b). In addition, immunoblotting revealed that WAT protein levels of mitochondrial chaperones and proteases, including LONP1, were higher in NCD-fed AdKO mice (iWAT: three- to fivefold; eWAT: four- to fivefold compared with the control group, Fig. 5a). Next, we investigated the mitochondrial stress response in AdKO and control mice fed an HFD for 4 weeks. AdKO mice showed increased WAT expression of UPRmt-associated genes involved in mitochondrial quality under HFD-fed conditions, and the increased protein levels of these genes in AdKO mice were also higher under HFD-fed conditions (iWAT: five- to 24-fold; eWAT: two- to 46-fold compared with the control group, ESM Fig. 7c, d and Fig. 5b). However, skeletal muscle and hepatic levels of these proteins were similar between AdKO and control mice under NCD- and HFD-fed conditions (ESM Fig. 8a, b). To evaluate a putative common mitochondrial stress response between genetic and pharmacological inhibition of adipocyte mitochondrial OxPhos function, we treated 3 T3-L1 adipocytes with doxycycline, which inhibits mitochondrial translation [13, 23]. Doxycycline statistically increased expression of Hspd1 mRNA and HSPD1 protein (ESM Fig. 7e and Fig. 5c), as well as the expression of LONP1 protein, in cultured adipocytes (Fig. 5c), which indicates that both genetic and pharmacological inhibition of mitochondrial OxPhos activated the adipose UPRmt.

Adipocyte-specific Crif1 knockout induces the UPRmt in adipose tissue. (a) Immunoblotting and band density of UPRmt proteins in iWAT (n = 3) and eWAT (n = 4) from 10-week-old NCD-fed Ctrl and AdKO mice. (b) Immunoblotting and band density of UPRmt proteins in iWAT and eWAT from Ctrl (n = 4) and AdKO mice (n = 5) fed an HFD for 4 weeks. (c) Immunoblotting and band density for UPRmt proteins in doxycycline (10 μl/ml or 20 μl/ml)-treated differentiated 3 T3-L1 adipocytes (n = 3; repeated twice using the same samples for the 10 μg/ml treatment). *p<0.05 vs Ctrl NCD (a) or Ctrl HFD (b) or None group (c) by Student’s t test. Ctrl, control; Doxy, doxycycline
Adipocyte-specific impairment of OxPhos function is associated with greater synthesis of adipo-mitokines in vivo
The transcriptional profile of adipocytes with impaired OxPhos is unknown. Therefore, we performed RNA sequencing of iWAT transcripts from control and AdKO mice fed an NCD. A large number of transcripts were altered in AdKO mice (ESM Fig. 9a; fold change increase or decrease (|FC|) >2; p<0.05). In particular, expression of major mitokines, such as Gdf15 and Fgf21, was much higher in the adipose tissue of AdKO mice compared with control mice (Fig. 6a and ESM Fig. 9b), as was the expression of metabolic pathway genes and genes involved in the phosphoinositol 3-kinase (PI3)–Akt and peroxisome proliferator-activated receptor signalling pathways (ESM Fig. 9c).

Adipocyte-specific Crif1 knockout induces production of adipocyte-derived mitokines in vivo and in vitro. (a) Heat map showing up- and downregulated genes in iWAT from NCD-fed Ctrl and AdKO mice at 10 weeks of age, with fold change values. The red text (Gdf15, Fgf21) indicates mitokines showing much higher fold change in AdKO mice among the secreted proteins. (b, c) Relative expression of mRNA encoding Gdf15 in (b) iWAT and (c) eWAT from 10-week-old Ctrl and AdKO mice consuming either an NCD or HFD (n = 4). The value was normalised to 18 s rRNA and expressed as fold change of the value in Ctrl extracts. (d, e) Relative protein expression of GDF15 (fold difference from Ctrl group) in (d) iWAT and (e) eWAT from 10-week-old mice consuming either an NCD (n = 3) or HFD (n = 4). (f, g) Relative mRNA expression of Fgf21 in (f) iWAT and (g) eWAT from 10-week-old Ctrl and AdKO mice consuming either an NCD or HFD (n = 4). The value was normalised to 18 s rRNA and expressed as a fold change of the value in Ctrl extracts. (h, i) Relative protein expression of FGF21 (fold difference from Ctrl group) in (h) iWAT and (i) eWAT from 10-week-old mice consuming either an NCD (n = 3) or HFD (n = 4). (j, k) Relative mRNA expression of Gdf15 and Fgf21 in the SVF and adipocytes isolated from the (j) iWAT and (k) eWAT of 10-week-old mice fed an NCD (n = 3). The value was normalised to 18 s rRNA and expressed as a fold change of the value in control adipocyte extracts. (l) Serum GDF15 and (m) serum FGF21 concentrations in 10-week-old Ctrl and AdKO mice fed an NCD (n = 3) or HFD (n = 4). *p<0.05 vs Ctrl group by Student’s t test (b–i, l, m). *p<0.05 vs Adipo Ctrl group by one-way ANOVA (j, k). Adipo Ctrl, control adipocytes; Ctrl, control
Next, we analysed iWAT and eWAT expression of these mitokines in control and AdKO mice under NCD-fed and HFD-fed conditions. Expression of Gdf15 mRNA and GDF15 protein were higher in both NCD- and HFD-fed AdKO mice, except for GDF15 protein levels in eWAT from NCD-fed mice (Fig. 6b–e). In addition, expression of iWAT and eWAT Fgf21 mRNA and FGF21 protein was significantly higher in AdKO mice than in controls fed either diet (Fig. 6f–i). To further evaluate cell type-specific gene expression in adipose tissue, we prepared adipocyte-enriched and SVFs from iWAT and eWAT. In both iWAT and eWAT, Gdf15 and Fgf21 transcript numbers increased in AdKO mice only in the adipocyte-enriched fraction, suggesting that impaired OxPhos increases expression of mitokines in adipocytes, but not in other adipose tissue cell types, including immune cells (Fig. 6j, k). In addition, pharmacological inhibition of OxPhos using doxycycline increased expression of mRNA encoding Gdf15 and Fgf21 in differentiated 3 T3-L1 adipocytes in a dose-dependent manner (ESM Fig. 10). Increased adipocyte mitokine expression probably contributed to high serum mitokine levels in AdKO mice, which were elevated under both NCD-fed and HFD-fed conditions (Fig. 6l, m). Taken together, these data suggest that genetic or pharmacological attenuation of adipocyte OxPhos promotes upregulation and secretion of several mitokines.
GDF15 and FGF21 attenuates progression of diet-induced obesity in AdKO mice
To elucidate the effects of GDF15 and FGF21 on energy metabolism and glucose homeostasis in AdKO mice, we generated AdKO mice with global Gdf15 deletion (AdGKO) or global Fgf21 deletion (AdFKO), both on a C57BL/6 background. We confirmed that GDF15 and FGF21 were not detected in the serum of AdGKO and AdFKO mice, respectively (ESM Fig. 11a, b). After consuming an HFD for 8 weeks, AdGKO and AdFKO mice exhibited significantly higher body mass gain than AdKO mice (Fig. 7a, d), accompanied by increased fat and liver masses (Fig. 7b, e). In addition, H&E staining and quantitative assessment of iWAT and eWAT adipocyte size revealed more pronounced adipocyte hypertrophy in AdGKO and AdFKO mice than in AdKO mice (Fig. 7c, f and ESM Fig. 11c, d). Hepatic fat accumulation was also much more pronounced in AdGKO and AdFKO mice (Fig. 7c, f). These findings suggest that mitokines regulate body weight and alleviate diet-induced obesity in AdKO mice, and may therefore be responsible for the protective effects of adipocyte-specific disruption of OxPhos in this context.

GDF15 and FGF21 contribute to weight gain control and protection from diet-induced obesity in AdKO mice. (a) Body mass of Ctrl, AdKO, and AdGKO mice that consumed an HFD from 6 to 14 weeks of age (n = 4). (b) Organ masses (normalised to body mass) of 14-week-old Ctrl, AdKO and AdGKO mice fed an HFD for 8 weeks (n = 4). (c) Representative images of H&E staining of iWAT, eWAT and liver sections from 14-week-old Ctrl, AdKO and AdGKO mice fed an HFD for 8 weeks (n = 4); scale bars, 50 μm. (d) Body mass of Ctrl, AdKO and AdFKO mice fed an HFD from 6 to 14 weeks of age (n = 4). (e) Organ masses (normalised to body mass) of 14-week-old Ctrl, AdKO and AdFKO mice fed an HFD for 8 weeks (n = 4). (f) Representative images of H&E staining of iWAT, eWAT and liver sections from 14-week-old Ctrl, AdKO and AdFKO mice fed an HFD for 8 weeks (n = 4); scale bars, 50 μm. *p<0.05 for AdKO vs Ctrl, or as shown; †p<0.05 for AdKO vs AdGKO; ¶p<0.05 for AdKO vs AdFKO, by one-way ANOVA. BW, body weight; Ctrl, control
GDF15 and FGF21 differentially regulate energy homeostasis in AdKO mice
Next, we sought to determine the effects of FGF21 and GDF15 on energy homeostasis and adipose immune cell populations in HFD-fed AdKO mice. Owing to the importance of adipose immune cells in whole-body energy homeostasis, we determined the effect of mitokines on the adipose immune cell population in HFD-fed AdKO mice using flow cytometry analysis. AdGKO mice had a larger population of M1 macrophages and a smaller population of M2 macrophages than AdKO mice (Fig. 8a and ESM Fig. 12a, c). Furthermore, EE (adjusted by total body mass) was significantly lower in AdGKO mice than in AdKO mice at night (Fig. 8b and ESM Fig. 13a), implying that adipocyte-derived GDF15 not only affected macrophage polarisation, but also enhanced EE, in AdKO mice. Moreover, induction of UCP1 that occurred secondary to decreased OxPhos function in AdKO mice (Fig. 4d) was significantly attenuated by global knockout of Gdf15 in AdKO mice (Fig. 8c). AdKO mice with global Fgf21 deletion had a higher iWAT and eWAT M1/M2 macrophage ratios than AdKO mice (Fig. 8d and ESM Fig. 12b, d). However, the increased EE (adjusted to total body mass) in AdKO mice was maintained in AdFKO mice, indicating that FGF21 did not regulate EE in AdKO mice (Fig. 8e and ESM Fig. 13b). Moreover, global Fgf21 deficiency did not affect adipose UCP1 expression in AdKO mice, with similar levels in AdKO and AdFKO mice (Fig. 8f), in contrast to the phenotype of AdGKO mice.

GDF15 and FGF21 play differential roles in regulating energy homeostasis in AdKO mice. (a) Representative flow cytometry plots of M1 (CD11c+CD206−) and M2 (CD11c−CD206+) macrophages within the total macrophage population in WAT SVFs of 14-week-old Ctrl, AdKO and AdGKO mice fed an HFD for 8 weeks (n = 3). Scatter plots and graphs of M1/M2 ratio are shown in ESM Fig. 12. (b) AUC for EE in 14-week-old Ctrl (n = 4), AdKO (n = 4) and AdGKO (n = 3) mice fed an HFD for 8 weeks. (c) Immunoblots showing UCP1 and β-actin and protein band density in WAT from 14-week-old Ctrl, AdKO and AdGKO mice fed an HFD for 8 weeks (n = 6). (d) Representative flow cytometry plots of M1 (CD11c+CD206−) and M2 (CD11c−CD206+) macrophages within the total macrophage population in WAT SVFs of 14-week-old Ctrl, AdKO and AdFKO mice fed an HFD for 8 weeks (n = 3). Scatter plots and graphs of M1/M2 ratio are shown in ESM Fig. 12. (e) AUC for EE in 14-week-old Ctrl (n = 4), AdKO (n = 5), and AdFKO (n = 3) mice fed an HFD for 8 weeks. (f) Immunoblots showing UCP1 and α-tubulin and protein band density in WAT from 14-week-old Ctrl, AdKO and AdFKO mice fed an HFD for 8 weeks (n = 6). (g) GTT and (h) ITT in 14-week-old Ctrl, AdKO and AdGKO mice fed an HFD for 8 weeks (n = 4). (i) GTT and (j) ITT in 14-week-old Ctrl, AdKO and AdFKO mice fed an HFD for 8 weeks (n = 4). *p<0.05 for AdKO vs Ctrl, or as shown; †p < 0.05 for AdKO vs AdGKO; ¶p<0.05 for AdKO vs AdFKO, by one-way ANOVA. Ctrl, control
Next, we used GTT and ITT to evaluate the effects of these mitokines on systemic glucose turnover and insulin sensitivity in HFD-fed AdKO mice. Despite greater body mass and adiposity in AdGKO mice, AdKO and AdGKO mice showed similar glucose tolerance (Fig. 8g), suggesting that improved glucose tolerance in AdKO mice was not mediated through GDF15. However, insulin stimulation did not as effectively stimulate glucose disposal in AdGKO mice as in AdKO mice, suggesting that upregulated GDF15 in AdKO mice contributed to improved insulin sensitivity under HFD-fed conditions (Fig. 8h). By contrast, AdFKO mice showed impaired glucose tolerance (Fig. 8i) and insulin sensitivity (Fig. 8j) compared with AdKO mice under HFD-fed conditions, suggesting that upregulated FGF21 in AdKO mice contributed to systemic glucose homeostasis and insulin sensitivity in the context of HFD. Although AdGKO and AdFKO mice fed an NCD had higher body weight (ESM Fig. 14a) and fat mass (ESM Fig. 14b) than AdKO mice, metabolic variables, including EE and glucose tolerance, were unchanged under NCD-fed conditions (ESM Fig. 14c, d). Taken together, these findings suggest that mitokines induced by the stress of impaired adipocyte OxPhos affect systemic energy metabolism and whole-body glucose homeostasis, which has a protective effect against diet-induced obesity.
Discussion
As the principal means of ATP production in eukaryotic cells, mitochondrial OxPhos is critical for normal glucose metabolism and systemic energy homeostasis. Mitochondrial OxPhos deficits can result from mitochondrial genetic disorders, but are also common in individuals with metabolic diseases such as obesity or type 2 diabetes [24]. Paradoxically, accumulating evidence also suggests that mitochondrial stress- or OxPhos dysfunction-induced UPRmt, which is evolutionarily conserved from worms to mammals [25, 26], has beneficial effects on whole-body metabolism [18, 19, 27]. However, most studies of this phenomenon have used lower organisms or global mouse knockout models, so the effects of this response in specific tissues remain incompletely understood. Therefore, we aimed to determine how adipose OxPhos function affects energy homeostasis in vivo. In the present study, we demonstrated that adipocyte-specific reduction in OxPhos function was associated with activation of local UPRmt and mitohormesis through increased transcription and secretion of FGF21 and GDF15, which are protective against the metabolic defects of diet-induced obesity.
In a prior study, we developed a mouse model with adipocyte-specific impairment in OxPhos function using Fabp4-Cre mice [28]. Unlike the AdKO mice, Crif1f/+Fabp4 mice exhibited adipose M1 macrophage-mediated inflammation and insulin resistance. This discrepancy between AdKO mice and Crif1f/+Fabp4 mice may be explained by inherent differences in the distribution and activity of Cre-recombinase driven by the Fabp4 or Adipoq promoters. Previous reports reveal that Fabp4 is expressed in macrophages [29] and the lymphatic system [30] in adult animals, and is detected within the embryo [31]; this may explain (in part) the difference between AdKO mice and Crif1f/+Fabp4 mice. Another mouse model of adipocyte mitochondrial dysfunction was also generated by crossing aP2-cre (also known as Fabp4-Cre) mice with Tfamf/f mice [6]. These mice had lower mtDNA copy number, but higher EE, and were protected against HFD-induced obesity and insulin resistance, similar to the phenotype of AdKO mice observed in the present study. By contrast, a study of adipocyte-specific Pgc-1β (also known as Ppargc1b)-deficient mice demonstrated that lower mitochondrial oxidative capacity in adipocytes is not sufficient for insulin resistance to develop, regardless of diet type [32].
Moreover, decreased white adipocyte OxPhos capacity appears to be a hallmark of obesity, although evidence of a causal role for mitochondrial dysfunction in obesity and metabolic diseases is lacking [33]. Furthermore, adipose tissue mitochondrial dysfunction can lead to a syndrome of lipodystrophy with insulin resistance and hepatic fat accumulation in mice [34]. These findings suggest that the relationship between mitochondrial OxPhos function and metabolic disease is complex, and is also likely to be context-specific. Therefore, the mechanisms by which tissue-specific OxPhos function affects whole-body metabolism remain unclear, but we have identified a causal link between adipose OxPhos function and systemic energy homeostasis in this study.
Mitochondrial dysfunction elicits a cellular stress response and resultant secretion of mitokines in various models and species [18, 35, 36]. UPRmt-associated secretory signals are proposed to promote longevity and improve health [37]. Recent studies show that activation of the UPRmt could be an important determinant of longevity in lower organisms such as Caenorhabditis elegans [37] and Drosophila melanogaster [25]. Knockdown of mitochondrial ribosomal protein S5-mediated induction of the UPRmt increases lifespan, accompanied by reduced \( \dot{V}{\mathrm{O}}_2 \), ATP content and citrate synthase activity in C. elegans. Redox signalling is also involved in activation of the UPRmt, which increases lifespan and preserves mitochondrial function in D. melanogaster. Although there is presently no evidence for a role of the UPRmt in longevity or metabolic diseases in mammals, mitokines such as FGF21 and GDF15 are not only useful diagnostic biomarkers for human mitochondrial diseases, but are also a potential therapeutic modality for metabolic diseases [18, 35, 36, 38, 39]. Moreover, Fgf21−/− and Gdf15−/− mice are prone to HFD -induced obesity, glucose intolerance, and hepatic and adipose inflammation [40, 41], suggesting an important role for these mitokines in whole-body metabolic homeostasis.
In the present study, we screened strong candidates for potential mitokine signals secreted by the adipose tissue of AdKO mice using RNA sequencing, and showed that mitokines play an important role in the phenotype associated with OxPhos dysfunction in AdKO mouse adipocytes by generating AdKO mice with global deletion of Gdf15 (AdGKO) or Fgf21 (AdFKO). To exclude the effects of GDF15 and FGF21 from other tissues, we used global Gdf15−/−or Fgf21−/−mice instead of adipose tissue-specific Gdf15- or Fgf21-deleted mice. Our data from AdGKO mice fed an HFD for 8 weeks revealed that long-term induction of GDF15 in AdKO mice attenuated progression of obesity in this context through increased EE. Our findings in AdFKO mice suggested that prolonged induction of FGF21 in AdKO mice did not affect EE, but remarkably ameliorated HFD-induced obesity and insulin resistance. However, further longer-term studies are warranted to determine whether the metabolic phenotypic effects of impaired OxPhos-mediated induction of the UPRmt are transient or persistent in mammals.
In humans, BAT activation protects against obesity and insulin resistance through non-shivering thermogenesis [42, 43]. The functional capacity and expression of UCP1 in BAT is impaired by chronic obesity in mice [44]. However, HFD- and thermoneutrality-induced whitening and dysfunction of BAT are likely to only modestly affect systemic glucose metabolism and EE. Consistent with this, surgical removal of BAT does not exacerbate diet-induced obesity or disrupt systemic glucose or lipid homeostasis in mice under limited thermal stress [45]. However, in the present study, AdKO mice showed decreased BAT UCP1 expression, but exhibited better glucose tolerance and higher EE in the context of HFD. Despite lower BAT UCP1 expression, the higher EE in AdKO mice may result from marked UCP1 induction in the iWAT and eWAT as well as from a higher concentration of mitokines. Consistent with this, ectopic WAT UCP1 expression improves glucose tolerance and insulin sensitivity in mice and rats [46, 47]. In addition, beige adipocytes are critically involved in the regulation of systemic glucose homeostasis and EE, as demonstrated by mice overexpressing PR domain-containing 16 in iWAT [47].
Taken together, our data demonstrate that AdKO mice show dual activation of cell-autonomous (chaperones and proteases) and non-cell-autonomous (mitokine) mechanisms in WAT. We have yet to confirm whether decreased OxPhos capacity directly induces secretion of adipo-mitokines. However, both AdGKO and AdFKO mice demonstrated a less pronounced iWAT UPRmt than AdKO mice under NCD-fed conditions, but the UPRmt was more marked than wild-type control mice (ESM Fig. 14e), indicating that the UPRmt, which was activated by mitochondrial stress in AdKO mice, was further increased by mitokine secretion. Although activation of the UPRmt and increased mito-adipokine secretion were identified, there were no differences in metabolic endpoints between AdKO, AdGKO, and AdFKO mice (ESM Fig. 14c, d) under NCD conditions, implying that a threshold concentration of mitokines may be required to regulate whole-body metabolism. Although these findings do not fully establish cause–effect relationships, we suggest that the adipose OxPhos function and mitohormesis from WAT can influence systemic glucose homeostasis and EE in pathologic states, such as diet-induced obesity. Metabolic dysfunction in Fgf21−/−and Gdf15−/−mice also supports our findings in the AdFKO and AdGKO mice used in the present study [40, 41]. Thus, we propose a novel role for the UPRmt and mitokine secretion in adipose tissue, which regulates both systemic glucose homeostasis and EE as part of an organismal adaptation to local mitochondrial stress. However, the human relevance of adipose mitochondrial OxPhos dysfunction and mitokine secretion in systemic energy metabolism needs to be clarified.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- AdFKO:
-
AdKO mice with global Fgf21 knockout
- AdGKO:
-
AdKO mice with global Gdf15 knockout
- AdKO:
-
Adipocyte-specific Crif1 knockout (mice)
- BAT:
-
Brown adipose tissue
- BN-PAGE:
-
Blue native-PAGE
- CLPP:
-
Caseinolytic mitochondrial matrix proteolytic subunit
- CRIF1:
-
Mitochondrial large ribosomal subunit protein
- DNAJA3:
-
DnaJ heat shock protein family (Hsp40) member A3
- EE:
-
Energy expenditure
- eWAT:
-
Epididymal white adipose tissue
- FGF21:
-
Fibroblast growth factor 21
- GDF15:
-
Growth differentiation factor 15
- HFD:
-
High-fat diet
- HSPD1:
-
Heat shock 60 kDa protein 1
- iWAT:
-
Inguinal WAT
- LONP1:
-
Lon peptidase 1
- NCD:
-
Normal chow diet
- NDUFA9:
-
NADH:ubiquinone oxidoreductase subunit A9
- NDUFB8:
-
NADH:ubiquinone oxidoreductase subunit B8
- OxPhos:
-
Oxidative phosphorylation
- SDHA:
-
Succinate dehydrogenase complex flavoprotein subunit A
- SVF:
-
Stromal vascular fraction
- UCP1:
-
Uncoupling protein 1
- UPRmt :
-
Mitochondrial unfolded protein response
- UQCRC2:
-
Ubiquinol–cytochrome c reductase core protein 2
- WAT:
-
White adipose tissue
References
Brand MD, Reynafarje B, Lehninger AL (1976) Stoichiometric relationship between energy-dependent proton ejection and electron transport in mitochondria. Proc Natl Acad Sci U S A 73(2):437–441. https://doi.org/10.1073/pnas.73.2.437
Perks KL, Ferreira N, Richman TR et al (2017) Adult-onset obesity is triggered by impaired mitochondrial gene expression. Sci Adv 3(8):e1700677. https://doi.org/10.1126/sciadv.1700677
Silva JP, Kohler M, Graff C et al (2000) Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet 26(3):336–340. https://doi.org/10.1038/81649
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med 350(7):664–671. https://doi.org/10.1056/NEJMoa031314
Dahlman I, Forsgren M, Sjogren A et al (2006) Downregulation of electron transport chain genes in visceral adipose tissue in type 2 diabetes independent of obesity and possibly involving tumor necrosis factor-alpha. Diabetes 55(6):1792–1799. https://doi.org/10.2337/db05-1421
Vernochet C, Mourier A, Bezy O et al (2012) Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab 16(6):765–776. https://doi.org/10.1016/j.cmet.2012.10.016
Bogacka I, Ukropcova B, McNeil M, Gimble JM, Smith SR (2005) Structural and functional consequences of mitochondrial biogenesis in human adipocytes in vitro. J Clin Endocrinol Metab 90(12):6650–6656. https://doi.org/10.1210/jc.2005-1024
Olswang Y, Cohen H, Papo O et al (2002) A mutation in the peroxisome proliferator-activated receptor gamma-binding site in the gene for the cytosolic form of phosphoenolpyruvate carboxykinase reduces adipose tissue size and fat content in mice. Proc Natl Acad Sci U S A 99(2):625–630. https://doi.org/10.1073/pnas.022616299
Fassina G, Dorigo P, Gaion RM (1974) Equilibrium between metabolic pathways producing energy: a key factor in regulating lipolysis. Pharmacol Res Commun 6(1):1–21. https://doi.org/10.1016/s0031-6989(74)80010-x
Koh EH, Park JY, Park HS et al (2007) Essential role of mitochondrial function in adiponectin synthesis in adipocytes. Diabetes 56(12):2973–2981. https://doi.org/10.2337/db07-0510
Quiros PM, Mottis A, Auwerx J (2016) Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol 17(4):213–226. https://doi.org/10.1038/nrm.2016.23
Yoneda T, Benedetti C, Urano F, Clark SG, Harding HP, Ron D (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 117(Pt 18):4055–4066. https://doi.org/10.1242/jcs.01275
Moullan N, Mouchiroud L, Wang X et al (2015) Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. https://doi.org/10.1016/j.celrep.2015.02.034
Durieux J, Wolff S, Dillin A (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144(1):79–91. https://doi.org/10.1016/j.cell.2010.12.016
Kim SJ, Kwon MC, Ryu MJ et al (2012) CRIF1 is essential for the synthesis and insertion of oxidative phosphorylation polypeptides in the mammalian mitochondrial membrane. Cell Metab 16(2):274–283. https://doi.org/10.1016/j.cmet.2012.06.012
Jung SB, Choi MJ, Ryu D et al (2018) Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nat Commun 9(1):1551. https://doi.org/10.1038/s41467-018-03998-z
Ryu MJ, Kim SJ, Choi MJ et al (2013) Mitochondrial oxidative phosphorylation reserve is required for hormone- and PPARgamma agonist-induced adipogenesis. Molecules and Cells 35(2):134–141. https://doi.org/10.1007/s10059-012-2257-1
Chung HK, Ryu D, Kim KS et al (2017) Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J Cell Biol 216(1):149–165. https://doi.org/10.1083/jcb.201607110
Bhaskaran S, Pharaoh G, Ranjit R et al (2018) Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep 19(3). https://doi.org/10.15252/embr.201745009
Lee HJ, Chung K, Lee H, Lee K, Lim JH, Song J (2011) Downregulation of mitochondrial lon protease impairs mitochondrial function and causes hepatic insulin resistance in human liver SK-HEP-1 cells. Diabetologia 54(6):1437–1446. https://doi.org/10.1007/s00125-011-2074-z
Segal KR, Landt M, Klein S (1996) Relationship between insulin sensitivity and plasma leptin concentration in lean and obese men. Diabetes 45(7):988–991. https://doi.org/10.2337/diab.45.7.988
Di Gregorio GB, Yao-Borengasser A, Rasouli N et al (2005) Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes 54(8):2305–2313. https://doi.org/10.2337/diabetes.54.8.2305
Quiros PM, Prado MA, Zamboni N et al (2017) Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216(7):2027–2045. https://doi.org/10.1083/jcb.201702058
Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT (2006) Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab 3(1):9–13. https://doi.org/10.1016/j.cmet.2005.12.001
Owusu-Ansah E, Song W, Perrimon N (2013) Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155(3):699–712. https://doi.org/10.1016/j.cell.2013.09.021
Wu Y, Williams EG, Dubuis S et al (2014) Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 158(6):1415–1430. https://doi.org/10.1016/j.cell.2014.07.039
Chen HS, Wu TE, Juan CC, Lin HD (2009) Myocardial heat shock protein 60 expression in insulin-resistant and diabetic rats. J Endocrinol 200(2):151–157. https://doi.org/10.1677/JOE-08-0387
Ryu MJ, Kim SJ, Kim YK et al (2013) Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PLoS Genet 9(3):e1003356. https://doi.org/10.1371/journal.pgen.1003356
Fu Y, Luo N, Lopes-Virella MF (2000) Oxidized LDL induces the expression of ALBP/aP2 mRNA and protein in human THP-1 macrophages. J Lipid Res 41(12):2017–2023
Ferrell RE, Kimak MA, Lawrence EC, Finegold DN (2008) Candidate gene analysis in primary lymphedema. Lymphat Res Biol 6(2):69–76. https://doi.org/10.1089/lrb.2007.1022
Urs S, Harrington A, Liaw L, Small D (2006) Selective expression of an aP2/fatty acid binding protein 4-Cre transgene in non-adipogenic tissues during embryonic development. Transgenic Res 15(5):647–653. https://doi.org/10.1007/s11248-006-9000-z
Enguix N, Pardo R, Gonzalez A et al (2013) Mice lacking PGC-1beta in adipose tissues reveal a dissociation between mitochondrial dysfunction and insulin resistance. Mol Metab 2(3):215–226. https://doi.org/10.1016/j.molmet.2013.05.004
Schottl T, Kappler L, Fromme T, Klingenspor M (2015) Limited OXPHOS capacity in white adipocytes is a hallmark of obesity in laboratory mice irrespective of the glucose tolerance status. Mol Metab 4(9):631–642. https://doi.org/10.1016/j.molmet.2015.07.001
Vernochet C, Damilano F, Mourier A et al (2014) Adipose tissue mitochondrial dysfunction triggers a lipodystrophic syndrome with insulin resistance, hepatosteatosis, and cardiovascular complications. FASEB J 28(10):4408–4419. https://doi.org/10.1096/fj.14-253971
Fujita Y, Ito M, Kojima T, Yatsuga S, Koga Y, Tanaka M (2015) GDF15 is a novel biomarker to evaluate efficacy of pyruvate therapy for mitochondrial diseases. Mitochondrion 20:34–42. https://doi.org/10.1016/j.mito.2014.10.006
Suomalainen A, Elo JM, Pietilainen KH et al (2011) FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol 10(9):806–818. https://doi.org/10.1016/S1474-4422(11)70155-7
Houtkooper RH, Mouchiroud L, Ryu D et al (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497(7450):451–457. https://doi.org/10.1038/nature12188
Chung HK, Kim JT, Kim HW et al (2017) GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Sci Rep 7(1):17238. https://doi.org/10.1038/s41598-017-17574-w
Kim KH, Jeong YT, Oh H et al (2013) Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19(1):83–92. https://doi.org/10.1038/nm.3014
Tran T, Yang J, Gardner J, Xiong Y (2018) GDF15 deficiency promotes high fat diet-induced obesity in mice. PLoS One 13(8):e0201584. https://doi.org/10.1371/journal.pone.0201584
Singhal G, Kumar G, Chan S et al (2018) Deficiency of fibroblast growth factor 21 (FGF21) promotes hepatocellular carcinoma (HCC) in mice on a long term obesogenic diet. Mol Metab 13:56–66. https://doi.org/10.1016/j.molmet.2018.03.002
Yoneshiro T, Aita S, Matsushita M et al (2013) Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest 123(8):3404–3408. https://doi.org/10.1172/JCI67803
Chondronikola M, Volpi E, Borsheim E et al (2014) Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes 63(12):4089–4099. https://doi.org/10.2337/db14-0746
Ohtomo T, Ino K, Miyashita R et al (2017) Chronic high-fat feeding impairs adaptive induction of mitochondrial fatty acid combustion-associated proteins in brown adipose tissue of mice. Biochem Biophys Rep 10:32–38. https://doi.org/10.1016/j.bbrep.2017.02.002
Grunewald ZI, Winn NC, Gastecki ML et al (2018) Removal of interscapular brown adipose tissue increases aortic stiffness despite normal systemic glucose metabolism in mice. Am J Physiol Regul Integr Comp Phys 314(4):R584–R597. https://doi.org/10.1152/ajpregu.00332.2017
Poher AL, Veyrat-Durebex C, Altirriba J et al (2015) Ectopic UCP1 overexpression in white adipose tissue improves insulin sensitivity in Lou/C rats, a model of obesity resistance. Diabetes 64(11):3700–3712. https://doi.org/10.2337/db15-0210
Seale P, Conroe HM, Estall J et al (2011) Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest 121(1):96–105. https://doi.org/10.1172/JCI44271
Acknowledgements
We are grateful to E. Rosen (Beth Israel Deaconess Medical Center, Boston) for providing the Adipoq-Cre transgenic mice, S-j Lee (Johns Hopkins University School of Medicine) for the Gdf15−/− mice, and N. Itoh (Kyoto University Graduate School of Pharmaceutical Sciences) for the Fgf21−/− mice.
Funding
This research was supported by the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT & Future Planning (No. NRF-2017R1E1A1A01075126), and the Global Research Laboratory (GRL) Program, through the NRF (No. NRF-2017K1A1A2013124). H-SY and JHL were also supported by the NRF (NRF-2015R1C1A1A01052432, NRF-2018R1C1B6004439 and NRF-2017R1A1A1A05001474, respectively).
Author information
Authors and Affiliations
Contributions
MJC, S-BJ, SEL and SGK performed data acquisition, data analysis and revised the article’s intellectual content. MJC, H-SY and MS made substantial contribution to conception and design of the study and drafting the work for important intellectual content. JHL and MJR contributed to the analysis and interpretation of data and critically revised the article. HKC, JYC, YKK, HJH, HK, HJK, C-HL and AM helped with the interpretation of data and contributed to drafting the article. MJC, H-SY and MS wrote the manuscript. All authors approved the final version of the manuscript. MS is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding authors
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM
(PDF 1394 kb)
Rights and permissions
About this article

Cite this article
Choi, M.J., Jung, SB., Lee, S.E. et al. An adipocyte-specific defect in oxidative phosphorylation increases systemic energy expenditure and protects against diet-induced obesity in mouse models. Diabetologia 63, 837–852 (2020). https://doi.org/10.1007/s00125-019-05082-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-05082-7