
Abstract
Aims/hypothesis
Oxidative stress is a promising target in diabetes-associated vasculopathies, with inhibitors of NADPH oxidases (NOX), in particular isoforms 1 and 4, shown to be safe in early clinical development. We have explored a highly relevant late-stage intervention protocol using the clinically most advanced compound, the NOX1/4 inhibitor GKT137831, to determine whether end-organ damage can be reversed/attenuated when GKT137831 is administered in the setting of established diabetic complications.
Methods
GKT137831 was administered at two doses, 30 mg kg−1 day−1 and 60 mg kg−1 day−1, to ApoE −/− mice 10 weeks after diabetes induction with streptozotocin (STZ), for a period of 10 weeks.
Results
Consistent with Nox4 −/− mouse data, GKT137831 was protective in a model of diabetic nephropathy at both the 30 mg kg−1 day−1 and 60 mg kg−1 day−1 doses, through suppression of proinflammatory and profibrotic processes. Conversely, in diabetic atherosclerosis, where Nox1 −/y and Nox4 −/− mice have yielded qualitatively opposing results, the net effect of pharmacological NOX1/4 inhibition was protection, albeit to a lower extent and only at the lower 30 mg kg−1 day−1 dose.
Conclusions/interpretation
As dose-dependent and tissue-specific effects of the dual NOX1/4 inhibitor GKT137831 were observed, it is critical to define in further studies the relative balance of inhibiting NOX4 vs NOX1 in the micro- and macrovasculature in diabetes.
Similar content being viewed by others


Introduction
The development of vascular complications, including atherosclerosis and kidney disease, is accelerated in diabetes [1]. Although a range of factors that contribute to the progression of diabetic complications have been identified, increased formation of reactive oxygen species (ROS), including by NADPH oxidase (NOX), is considered critically important. A major source of ROS is derived from members of the NOX family [2]. Three primary NOX isoforms have been identified in the mouse, NOX1, NOX2 and NOX4, and are considered to be relevant to vascular and renal disease [3, 4]. In the setting of diabetes, NOX1 has been shown to exert deleterious effects in retinopathy and macrovascular disease, whereas NOX4 has been shown to play a critical role in the development of diabetic nephropathy. Global Nox1 deletion attenuated atherosclerosis in diabetes [5] and genetic deletion of Nox4 globally and specifically in podocytes attenuated the development of kidney disease [6].
Therapeutic targeting of NOX-derived ROS had limited success initially, largely because of the lack of specific inhibitors. Early studies with apocynin and diphenyleneiodonium (DPI) demonstrated reductions in ROS production and attenuation of disease [7, 8]. However, both agents were shown to have significant off-target effects, in part because they are not specific NOX inhibitors [9, 10]. More recently, specific NOX inhibitors have been developed, including the NOX1/4 inhibitor GKT137831 (Genkyotex, Geneva, Switzerland). This compound does not affect NOX2-mediated phagocyte function and thus appears to be superior to other NOX/ROS inhibitors [11]. Previously, our group demonstrated that administration of GKT137831 to diabetic ApoE −/− mice attenuated the development of diabetes-associated atherosclerosis and diabetes-associated kidney disease [5, 6, 12]. We hypothesised that administration of GKT137831 in a model of diabetic macro- and microvascular disease would attenuate or even reverse disease progression. In these studies, GKT137831 was administered as a preventive intervention, starting immediately after diabetes induction with streptozotocin (STZ). A clinically more relevant question is whether NOX1/4 inhibition can arrest, attenuate or even reverse end-organ injury once disease progression is established.
Therefore, the overall aim of the current study was to explore if pharmacological inhibition of NOX1 and NOX4 in mice with established diabetic complications, including both macrovascular and renal disease, could attenuate or indeed reverse disease progression. We used the STZ-diabetes model in ApoE −/− mice and two doses of the specific NOX1 and NOX4 inhibitor GKT137831 administered in a setting of established diabetic nephropathy and atherosclerosis.
Methods
More detailed methods can be found in the electronic supplementary material (ESM).
Animal model
All animals were housed at the Precinct Animal Centre, Baker IDI, with all animal experiments being conducted in accordance to the principles and guidelines devised by the Alfred Medical Research & Education Precinct (AMREP) Animal Ethics Committee under the guidelines laid down by the National Health and Medical Research Council of Australia. The animals had unrestricted access to water and feed and were maintained on a 12 h light and dark cycle in a pathogen-free environment on standard mouse chow (Specialty Feeds, Glen Forrest, WA, Australia).
Male ApoE −/− mice on the C57 background, at 6 weeks of age were rendered diabetic by five daily i.p. injections of STZ (Sigma-Aldich, St Louis, MO, USA) at a dose of 55 mg/kg in citrate buffer [5, 13, 14]. Mice were excluded from the study if they had a blood glucose reading below 15 mmol/l 1 week after diabetes induction. At 19 weeks post induction of diabetes, systolic blood pressure was assessed by a non-invasive tail cuff system in conscious mice [15]. At 10 and 20 weeks after induction of diabetes, mice were placed individually into metabolic cages (Iffa Credo, L’Arbresele, France) for 24 h urine collection [6]. After 20 weeks, the animals were anaesthetised by i.p. injections of sodium pentobarbitone (Euthatal, 100 mg/kg body weight; Sigma-Aldrich, Castle Hill, NSW, Australia). HbA1c was measured in erythrocyte lysates by HPLC (Bio-Rad, Philadelphia, PA, USA). Plasma glucose, cholesterol and triacylglycerol concentrations were measured using enzymatic assays (Beckman Coulter Diagnostics, Gladsville, NSW, Australia). Organs were dissected and weights obtained before being either snap frozen in liquid nitrogen or fixed in buffered formalin (10% vol./vol.).
NOX inhibition
GKT137831 is a specific inhibitor of both NOX1 and NOX4 [16, 17]. GKT137831 was administered by oral gavage as a delayed intervention at a dose of 30 mg kg−1 day−1 or 60 mg kg−1 day−1 from week 10 to week 20 of diabetes. All animals were randomly assigned to treatment groups.
Atherosclerotic plaque area quantification
Assessment of plaque area was undertaken using en face analysis after being cleaned and stained with Sudan IV, as previously described, in a blinded fashion [18].
Renal function variables
Urinary albumin concentration was measured at 10 and 20 weeks after the induction of diabetes, using a mouse albumin ELISA (Bethyl Laboratories, Montgomery, TX, USA) [6, 19]. Urinary and serum creatinine concentrations were measured by HPLC [19, 20]. The urinary albumin/creatinine ratio (ACR) and creatinine clearance were calculated.
Quantitative RT-PCR
Total RNA was extracted from aorta and kidneys with DNA-free RNA being reverse transcribed into cDNA [5, 6]. Expression of genes encoding members of the NOX family, Nox1, Nox2 (also known as Cybb), Nox4 and p47phox (also known as Ncf1), proinflammatory and profibrotic pathways, including Tnfα (also known as Tnf), Nfκ p65 (also known as Rela), Mcp-1 (also known as Ccl2), Vcam-1 (also known as Vcam1), Ctgf, fibronectin and collagen IV were analysed by quantitative RT-PCR using the Taqman system (ABI Prism 7500; Perkin-Elmer, Foster City, CA, USA). The primer and probe sequences have been published previously [19,20,21,22,23].
Measurements of ROS and H2O2
Tissue ROS production was measured in 10 mm segments of aorta and renal cortex using L-012 (Wako Chemicals, Richmond, VA, USA) at a concentration of 100 μmol/l, in the dark, after incubation at 37°C for 15 min and read on a fluorescence microplate reader at 400 nm. The generation of H2O2 was measured in 10 mm segments of aorta and renal cortex samples by Amplex Red, using a commercial kit (Molecular Probes, Eugene, OR, USA) [5]. Cytosolic and mitochondrial extracts were isolated as previously described [24]. Fluorescence intensity was measured on a microplate at an excitation wavelength of 544 nm and an emission wavelength of 590 nm.
Immunohistochemistry
Paraffin sections (4 μm) of kidney were used for histological stains using periodic acid–Schiff’s reagent to assess mesangial expansion, as previously described [21, 25]. Immunohistochemical stains were performed on paraffin sections (4 μm) of aorta and kidney for nitrotyrosine, a marker of oxidative stress (rabbit polyclonal; Millipore, Billerica, MA, USA; 1:100), fibronectin, a marker of fibrosis (rabbit polyclonal; Abcam, Cambridge, AM USA; 1:200) and F4/80, a marker of macrophages (rat monoclonal; Abcam, Cambridge, AM, USA; 1:100) as previously described, and analysed in a blinded fashion [5, 26, 27].
MCP-1, TNF-α, IL1-β, TGF-β, VEGF and MIF measurements
Levels of aortic and/or kidney monocyte chemoattractant protein 1 (MCP-1), TNF-α, IL1-β, TGF-β, vascular endothelial growth factor (VEGF) and macrophage inhibitory factor (MIF) were measured as previously described [28]. In brief, whole-protein preparations were obtained [6] and measured following the manufacturer’s instructions (MCP-1, TNF-α, TGF-β, IL1-β and VEGF, R&D Systems, Minneapolis, MN, USA; MIF, Wuhan USCN, China).
Statistical analysis
Data were analysed for normality using the Shapiro–Wilk test before being analysed by either a one- or two-way ANOVA using SPSS Statistics version 20 (IBM, Armonk, NY, USA) with a least significant difference (LSD) post hoc test for multiple comparisons of the means. A p value <0.05 was considered statistically significant. Results are shown as mean ± SEM unless otherwise specified.
Results
Delayed NOX1/4 inhibition reverses urinary albumin excretion in diabetic mice
We tested if delayed administration of GKT137831 in mice with established diabetic complications including atherosclerosis and renal disease could attenuate end-organ injury. After 20 weeks of diabetes, untreated diabetic ApoE −/− mice gained less body weight than their untreated control counterparts (Table 1). Additionally, untreated diabetic mice had significant elevations in blood glucose, HbA1c, cholesterol, triacylglycerol and LDL-cholesterol (Table 1). Administration of GKT137831 from week 10 to 20 of diabetes at both doses did not affect body weight, blood glucose, HbA1c or circulating lipid concentrations (Table 1). Interestingly, administration of GKT137831 at the lower 30 mg kg−1 day−1 dose significantly reduced total kidney/body weight ratio compared with untreated ApoE −/− diabetic mice, but was ineffective at the higher 60 mg kg−1 day−1 dose (Table 1).
Albuminuria and urinary ACR, key functional markers of diabetic kidney disease, were significantly elevated in untreated diabetic ApoE −/− mice after 10 and 20 weeks of diabetes compared with untreated control ApoE −/− mice. Delayed treatment of diabetic ApoE −/− mice with both the 30 mg kg−1 day−1 and 60 mg kg−1 day−1 GKT137831 doses from week 10 of diabetes onwards was associated with reduced albuminuria (Fig. 1a, b).

There was a significant increase in urinary albumin excretion and ACR after 10 weeks of diabetes (a, b). After an additional 10 weeks of diabetes, untreated mice had a persistent increase in urinary albumin concentration (a) and ACR (b). In contrast, mice that received GKT137831 at the 30 mg kg−1 day−1 and the 60 mg kg−1 day−1 dose from week 10 to 20 of diabetes had attenuated urinary albumin concentrations (a) and ACR (b). Data are mean ± SEM (n = 8–10 per group); * p < 0.05 vs untreated ApoE −/− control mice; † p < 0.05 vs untreated ApoE −/− diabetic mice. Cont, control; Diab, diabetic; UT, untreated; wks, weeks
Delayed inhibition of NOX1/4 attenuates renal NOX isoform gene expression and markers of ROS in diabetes
After 20 weeks of diabetes, ApoE −/− mice demonstrated a significant increase in the gene expression of Nox1, Nox4 and p47phox in the renal cortex, with no change in the expression of Nox2 (Table 2). Administration of GKT137831 at the 30 mg kg−1 day−1 dose to diabetic ApoE −/− mice reduced the diabetes-induced increase in the gene expression of both Nox1 and Nox4, while the 60 mg kg−1 day−1 dose attenuated Nox4 and Nox2 gene expression (Table 2). Glomerular nitrotyrosine accumulation was significantly increased in untreated diabetic ApoE −/− mice after 20 weeks of diabetes, and this was not significantly altered in response to GKT137831 administration from week 10 to 20 of diabetes (Fig. 2a, c). Renal cortex H2O2 was significantly reduced in untreated diabetic ApoE −/− mice compared with untreated ApoE −/− controls, while measurements of ROS production demonstrated no change when comparing untreated ApoE −/− diabetic with ApoE −/− control mice (Fig. 2b, d). Administration of GKT137831 at both 30 mg kg−1 day−1 and 60 mg kg−1 day−1 did not have any effect on renal cortex H2O2 or ROS production (Fig. 2b, d).

Glomerular nitrotyrosine concentration was increased after 20 weeks of diabetes, and this was unaffected by delayed intervention treatment with GKT137831 at both doses (30 mg kg−1 day−1 and 60 mg kg−1 day−1) (a, c). Renal cortex H2O2 production was significantly lower in untreated diabetic ApoE −/− mice compared with untreated ApoE −/− control mice and was unaffected by GKT137831 administration (b). Renal cortex ROS production was unaffected in response to both diabetes and administration of GKT137831 in the setting of diabetes (d). Photomicrographs of nitrotyrosine immunohistochemistry were taken at ×40 magnification (c). Data are mean ± SEM (n = 8–10 per group); * p < 0.05 compared with untreated ApoE −/− control mice; † p < 0.05 compared with untreated ApoE −/− diabetic mice. Cont, control; Diab, diabetic; GKT, GKT137831; NT, nitrotyrosine; RLU, relative light units; 30 mg GKT, 30 mg kg−1 day−1; 60 mg GKT, 60 mg kg−1 day−1
Delayed inhibition of NOX1/4 attenuates renal inflammation and renal fibrosis
The diabetes-induced increase in mesangial expansion in untreated diabetic ApoE −/− mice after 20 weeks of diabetes was significantly attenuated by both doses of GKT137831, with the higher dose leading to a mesangial area approaching that seen in non-diabetic ApoE −/− mice (Fig. 3a, b). Diabetes induced significant increases in protein concentrations of the proinflammatory factors MCP-1 and TNF-α and the profibrotic factors VEGF and TGF-β in the renal cortex (Fig. 3c, d, f, g). Furthermore, there were significant increases in the expression of the collagen IV gene and glomerular fibronectin protein staining in untreated ApoE −/− diabetic mice (Fig. 3e, h). Delayed administration of GKT137831 at both doses significantly attenuated the diabetes-induced increase in MCP-1, TNF-α, VEGF and TGF-β concentrations, with the higher dose normalising protein concentrations to levels similar to those of the untreated non-diabetic ApoE −/− mice (Fig. 3c, d, f, g). Glomerular fibronectin protein levels were significantly reduced by the high dose of GKT137831 (60 mg kg−1 day−1) and non-significantly reduced at the low dose (30 mg kg−1 day−1) compared with untreated diabetic ApoE −/− mice (Fig. 3h, i).

Mesangial expansion and renal cortical MCP-1 concentrations were increased after 20 weeks of diabetes, with increases attenuated with GKT137831 administration at both doses (30 mg kg−1 day−1 and 60 mg kg−1 day−1) (a–c). The diabetes-induced increase in renal collagen IV gene expression was unaffected by GKT137831 administration (e). However, administration of GKT137831 at both doses significantly reduced the diabetes-induced increase in TNF-α (d), VEGF (f) and TGF-β (g) concentrations. Administration of GKT137831 at the higher dose of 60 mg kg−1 day−1 attenuated the diabetes-induced increase in glomerular fibronectin accumulation (h, i). Photomicrographs of mesangial area (b) and fibronectin immunohistochemistry (i) were taken at ×40 magnification. Data are shown as mean ± SEM (n = 8–10 per group); * p < 0.05 vs untreated control ApoE −/− mice; † p < 0.05 vs untreated diabetic ApoE −/− mice. Cont, control; Diab, diabetic; GKT, GKT137831; 30 mg GKT, 30 mg kg−1 day−1; 60 mg GKT, 60 mg kg−1 day−1
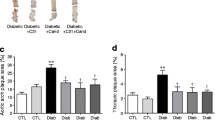
Delayed NOX1/4 inhibition demonstrates a dose-dependent anti-atherosclerotic effect in diabetes
After 20 weeks of diabetes, untreated diabetic ApoE −/− mice demonstrated a significant increase in plaque area compared with untreated ApoE −/− control mice (Fig. 4a, b, ESM Fig. 1). Administration of GKT137831 to ApoE −/− mice from week 10 until week 20 of diabetes at a dose of 30 mg kg−1 day−1 significantly attenuated the development of atherosclerosis compared with untreated diabetic ApoE −/− mice, resulting in a 30% reduction in aortic arch and total aortic plaque area (Fig. 4a, b). However, the effect was less pronounced than when the compound was given in a preventive manner (Fig. 4a, b). Interestingly, at the higher dose of 60 mg kg−1 day−1, GKT137831 was ineffective at preventing further development of atherosclerosis in the aortic arch and total aorta compared with untreated diabetic ApoE −/− mice (Fig. 4a, b).

There were significant increases in plaque area in the total aorta and aortic arch after 10 and 20 weeks of diabetes (a, b). Administration of GKT137831 as a delayed intervention (from weeks 10 to 20 of diabetes) at the dose of 30 mg kg−1 day−1 delayed the progression of atherosclerosis. However, the higher dose of 60 mg kg−1 day−1 GKT137831 did not alter atherosclerosis development. Data are shown as mean ± SEM (n = 8–10 per group); * p < 0.05 vs ApoE −/− untreated control mice; † p < 0.05 vs ApoE −/− untreated diabetic mice. Cont, control; Diab, diabetic; UT, untreated; wks, weeks
NOX1/4 inhibition modulates markers of ROS and expression of NOX isoforms in the aorta of diabetic mice
After 20 weeks of diabetes, untreated diabetic ApoE −/− mice demonstrated a significant increase in the aortic gene expression of Nox1, Nox2 and the cytosolic subunit p47phox, with a significant reduction in Nox4 gene expression (Table 3). Upon treatment with GKT137831 at the higher dose of 60 mg kg−1 day−1 there was a significant decrease in the gene expression of Nox1, Nox2, Nox4 and p47phox. At the lower dose of 30 mg kg−1 day−1, GKT137831 only resulted in a decrease in Nox2 and p47phox gene expression, with no change in gene expression of Nox1 (Table 3). Administration of GKT137831 at the lower dose of 30 mg kg−1 day−1 significantly increased the gene expression of Nox4 in the aorta compared with untreated diabetic ApoE −/− mice (Table 3). Nitrotyrosine was increased in the aorta of untreated diabetic ApoE −/− mice compared with control ApoE −/− mice (Fig. 5a). Administration of GKT137831 at both doses resulted in a significant reduction in aortic nitrotyrosine accumulation in the diabetic mice (Fig. 5a). Measurements of aortic H2O2 production identified a significant reduction in H2O2 production in untreated ApoE −/− diabetic mice compared with untreated control ApoE −/− mice. Administration of GKT137831 at 30 mg kg−1 day−1 significantly increased H2O2 production compared with untreated diabetic ApoE −/− mice (Fig. 5b). GKT137831 administration at the higher dose of 60 mg kg−1 day−1 had no effect on H2O2 production (Fig. 5b). In turn, measurements of aortic ROS production identified a non-significant increase in ROS production in untreated ApoE −/− diabetic mice, which was attenuated with administration of GKT137831 at 60 mg kg−1 day−1 but not at 30 mg kg−1 day−1 (Fig. 5d).

Aortic nitrotyrosine accumulation was increased after 20 weeks of diabetes. Treatment with GKT137831 at both doses (30 mg kg−1 day−1 and 60 mg kg−1 day−1) from week 10 to 20 attenuated this variable (a, c). Aortic H2O2 production was significantly reduced in untreated diabetic mice and was significantly restored above control levels with GKT137831 at 30 mg kg−1 day−1 but not 60 mg kg−1 day−1 (b). Aortic ROS production was largely unchanged across all groups except in the diabetic ApoE −/− mice treated with GKT137831 at the higher dose (d). Photomicrographs of nitrotyrosine immunohistochemistry were taken at ×40 magnification (c). Data are mean ± SEM (n = 8–10 per group); * p < 0.05 compared with untreated ApoE −/− control mice; † p < 0.05 compared with untreated ApoE −/− diabetic mice. Cont, control; Diab, diabetic; GKT, GKT137831; NT, nitrotyrosine; RLU, relative light units; 30 mg GKT, 30 mg kg−1 day−1; 60 mg GKT, 60 mg kg−1 day−1
Aortic inflammation and chemotaxis is reduced in response to pharmacological inhibition of NOX1/4 in diabetes
After 20 weeks of diabetes, ApoE −/− mice demonstrated a significant increase in the protein levels of TNF-α, IL1-β and MCP-1 (Fig. 6a–c). Measurements of the proinflammatory and pro-atherogenic MIF in aortic homogenates identified a significant diabetes-induced increase (Fig. 6d), which correlated with an increase in macrophage accumulation within the aortic wall (Fig. 6e, f). Delayed administration of GKT137831 significantly reduced MCP-1 protein concentration only at the lower dose of 30 mg kg−1 day−1 (Fig. 6c). Treatment with the higher dose of 60 mg kg−1 day−1 was associated with elevations in protein levels of TNF-α and IL-1β (Fig. 6a, b). GKT137831 administration significantly elevated MIF concentration at the higher dose (60 mg kg−1 day−1) with a non-significant reduction when administered at the lower dose (30 mg kg−1 day−1; Fig. 6d). Vascular macrophage accumulation within the aortic wall was non-significantly reduced with the lower dose of GKT137831 (30 mg kg−1 day−1), but no clear effect on vascular macrophage accumulation was seen with the higher dose of 60 mg kg−1 day−1 (Fig. 6e, f).

Aortic TNF-α, IL-1β, MCP-1 and MIF protein concentrations were increased in diabetic mice. GKT137831 administration at the low dose (30 mg kg−1 day−1) non-significantly reduced the concentration of TNF-α, IL-1β and MIF, and significantly reduced MCP-1 (a–d). In contrast, GKT137831 administration at the higher dose (60 mg kg−1 day−1) significantly increased the concentration of TNF-α, IL-1β and MIF, with no effect on MCP-1 (a–d). Similarly, macrophage accumulation was increased in response to diabetes, but GKT137831 did not alter macrophage accumulation (e, f). Photomicrographs of F4/80 immunohistochemistry were taken at ×40 magnification (f). Data are mean ± SEM (n = 8–10 per group); * p < 0.05 compared with untreated ApoE −/− control mice; † p < 0.05 compared with untreated ApoE −/− diabetic mice; ‡ p < 0.05 compared with ApoE −/− diabetic mice treated with 30 mg kg−1 day−1 GKT137831. Cont, control; Diab, diabetic; GKT, GKT137831; 30 mg GKT, 30 mg kg−1 day−1; 60 mg GKT, 60 mg kg−1 day−1
Discussion
There is increasing evidence that NOX-derived ROS formation plays a key role in the development and progression of micro- and macrovascular complications, in particular in the context of diabetes. We have previously shown that global deletion of Nox1 attenuated diabetes-associated atherosclerosis in a shorter-duration model of 10 weeks of diabetes via effects on inflammation and plaque remodelling. Preventive treatment with a novel NOX1/4 inhibitor GKT137831 in diabetic ApoE −/− mice similarly attenuated plaque development in association with increased plaque stability, as assessed by a reduced number of necrotic cores, attenuated macrophage accumulation and reduced vascular inflammation [5, 12]. Moreover, recently we have identified that NOX4 plays a protective role in diabetes-associated atherosclerosis [28].
The effects of NOX1/4 inhibition were different in the kidney, further emphasising tissue-specific effects of NOX isoform inhibition. We have previously shown, using global Nox4-knockout mice, that deletion of NOX4 affords renoprotective effects in diabetic kidney disease. In addition, podocyte-specific Nox4 deletion was shown to be renoprotective in diabetic kidney disease [6, 29], whereas Nox1 deletion had no impact [6].
Furthermore, NOX1/4 inhibition with GKT137831 has shown renoprotective effects including reduction in albuminuria and attenuation of renal structural injury and inflammation after 20 weeks of treatment [6]. Indeed, the preventive renoprotective effect of GKT137831 has been shown in at least three different animal models of diabetic nephropathy, including in ApoE −/− mice with STZ-induced diabetes [25], in the Akita mouse [30] and in the OVE26 mouse [31], as reflected by reduced albuminuria, renal fibrosis and inflammation.
Here, we report that delayed intervention with two doses of GKT137831 attenuates established renal disease in diabetes as assessed by lower albuminuria, a decrease in renal fibrosis and a reduction in renal inflammation. Indeed, delayed intervention with GKT137831 reversed the overt albuminuria observed after 10 weeks of diabetes by at least 50% by the end of the study. Renal Nox4 gene expression was significantly reduced with both doses of GKT137831 and this may reflect reduced renal injury. Similar effects were recently reported in microglial and Müller cells exposed to hypoxia and treated with GKT137831 [32]. It is therefore possible that reduced expression and enzyme activity both contribute to the pharmacological effects of NOX inhibitors. Furthermore, the higher dose of GKT137831 appeared to have a more potent effect on some of the renal variables examined, further supporting the deleterious role of NOX4 in diabetic nephropathy.
To assess the vasculoprotective effects of pharmacological NOX1/4 inhibition in a clinically more relevant context, we examined the impact of delayed intervention with the NOX1/4 inhibitor GKT137831 in established disease. Indeed, a dose of 30 mg kg−1 day−1 significantly attenuated atherosclerosis development in mice with established diabetic vascular disease. However, the higher dose of 60 mg kg−1 day−1 failed to reduce plaque area. At this dose, it is postulated that GKT137831 may have a more profound effect on vascular NOX4 activity, thereby offsetting the beneficial effects conferred by NOX1 inhibition.
Interestingly, delayed intervention with both doses of GKT137831 was associated with reduced aortic nitrotyrosine accumulation, consistent with a beneficial effect of vascular NOX1 inhibition. Interestingly, Nox4 expression was already reduced in the aorta of diabetic ApoE −/− mice. The lower dose of 30 mg kg−1 day−1 increased Nox4 expression, whereas the higher dose of 60 mg kg−1 day−1 had no effect. It is unclear if these differences contribute to the lack of atheroprotection seen with the higher dose. These findings are consistent with recent studies that have observed that NOX4 is atheroprotective [28]. It is, however, important to interpret gene expression results with caution, as Nox1 and Nox2 are mainly regulated at the post-translational level. The atheroprotective role of NOX4 can potentially explain the lack of atheroprotection at the high dose of GKT137831. Also, the diseased aorta undergoes substantial remodelling in this long-term model, with atherosclerotic plaques covering close to 30% of the luminal area. Therefore, tissue heterogeneity and increased acellular material inevitably reduce the ability to interpret these gene expression findings.
The renal cortex of untreated diabetic mice showed a depletion of H2O2. This finding has been previously reported in the diabetic kidney [33], although this phenomenon remains to be fully explained. Reductions in H2O2 levels most likely arise through reduced production by H2O2 generators such as NOX4, reduced dismutation from superoxide by the superoxide dismutase (SOD) enzymes, or increased removal by the peroxidase enzymes, of which there are several. The fact that Nox4 expression increased in the diabetic ApoE −/− kidney in this study may seem contradictory. However, two reports claim that NOX4 contributes to the generation of superoxide under some disease conditions [34, 35], highlighting the important nature of changes in ROS production between health and disease. H2O2 levels were also observed to increase in the aorta after 30 mg kg−1 day−1 GKT137831 treatment. One possible explanation is that the activity of peroxidases is reduced by NOX1/4 inhibition in the diabetic aorta, resulting in a lower rate of H2O2 removal to water. Furthermore, the generation of H2O2 occurs via multiple sources, not only via dismutation of superoxide by the SOD enzymes. Therefore, one cannot assume that an increase in superoxide would always lead to an increase in H2O2 as this reaction also relies on the activity of various SOD enzymes, which are altered in the diabetic milieu [36].
These results demonstrate that blocking NOX1/4 in established micro-and macrovascular complications of diabetes can provide concomitant athero- and renoprotection in diabetes. Furthermore, our results suggest that there are tissue- and dose-dependent effects of GKT137831 that need to be further explored. The degree of relative inhibition of NOX4 vs NOX1 appears to be particularly important in the macrovasculature, although this issue does not appear as important in the kidney.
Preliminary results of a short-term phase 2a clinical trial evaluating the safety and efficacy of GKT137831 in patients with diabetic kidney disease have been reported [37]. This report indicates that GKT137831 was very well tolerated and showed pharmacodynamic activity on markers of inflammation and liver injury. However, GKT137831 treatment for 12 weeks did not improve renal outcomes including albuminuria. While the reported safety and pharmacodynamic results appear encouraging, it will be important to assess these clinical results further and to determine specifically if treatment duration, dose, concomitant medications or certain characteristics of the patient population might have played a role.
Taken together, the available data suggest that selective inhibition of NOX1 may be particularly effective to specifically prevent diabetes-induced vascular disease in conduit arteries and the retina [32]. NOX inhibitors with a broader selectivity profile, including NOX1, NOX4 and, possibly, NOX5 may represent more attractive therapeutic options in patients with associated kidney disease. Therefore, NOX inhibitors with specific selectivity profiles should be evaluated in carefully defined patient populations. Given the dose- and tissue-specific effects of the dual NOX1/4 inhibitor GKT137831, future studies need to define the relative balance of NOX1 vs NOX4 inhibition in both the micro- and macrovasculature in diabetes. Any clinical study evaluating an agent such as GKT137831 should include concomitant assessment of both renal and cardiovascular endpoints.
Abbreviations
- ACR:
-
Albumin/creatinine ratio
- MCP-1:
-
Monocyte chemoattractant protein 1
- MIF:
-
Macrophage inhibitory factor
- NOX:
-
NADPH oxidase
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- STZ:
-
Streptozotocin
- VEGF:
-
Vascular endothelial growth factor
References
Shaw JE, Sicree RA, Zimmet PZ (2010) Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 87:4–14
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Brandes RP, Weissmann N, Schroder K (2010) NADPH oxidases in cardiovascular disease. Free Radic Biol Med 49:687–706
Lassegue B, Griendling KK (2010) NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30:653–661
Gray SP, Di Marco E, Okabe J et al (2013) NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127:1888–1902
Jha JC, Gray SP, Barit D et al (2014) Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J Am Soc Nephrol 25:1237–1254
Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ (1994) Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 11:95–102
Ferreira Mendes A, Pato Carvalho A, Margarida Caramona M, Celeste Lopes M (2001) Diphenyleneiodonium inhibits NF-κB activation and iNOS expression induced by IL-1β: involvement of reactive oxygen species. Mediat Inflamm 10:209–215
Heumuller S, Wind S, Barbosa-Sicard E et al (2008) Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 51:211–217
Riganti C, Gazzano E, Polimeni M, Costamagna C, Bosia A, Ghigo D (2004) Diphenyleneiodonium inhibits the cell redox metabolism and induces oxidative stress. J Biol Chem 279:47726–47731
Casas AI, Dao VT, Daiber A et al (2015) Reactive oxygen-related diseases: therapeutic targets and emerging clinical indications. Antioxid Redox Signal 23:1171–1185
Di Marco E, Gray SP, Chew P et al (2014) Pharmacological inhibition of NOX reduces atherosclerotic lesions, vascular ROS and immune-inflammatory responses in diabetic Apoe−/− mice. Diabetologia 57:633–642
Gavazzi G, Banfi B, Deffert C et al (2006) Decreased blood pressure in NOX1-deficient mice. FEBS Lett 580:497–504
Kleinschnitz C, Grund H, Wingler K, et al. (2010) Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol 8:e1000479
Krege JH, Hodgin JB, Hagaman JR, Smithies O (1995) A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 25:1111–1115
Jiang JX, Chen X, Serizawa N et al (2012) Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med 53:289–296
Aoyama T, Paik YH, Watanabe S et al (2012) Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 56:2316–2327
Candido R, Allen TJ, Lassila M et al (2004) Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation 109:1536–1542
Watson AM, Li J, Schumacher C et al (2010) The endothelin receptor antagonist avosentan ameliorates nephropathy and atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 53:192–203
Watson AM, Gray SP, Jiaze L et al (2012) Alagebrium reduces glomerular fibrogenesis and inflammation beyond preventing RAGE activation in diabetic apolipoprotein E knockout mice. Diabetes 61:2105–2113
Chew P, Yuen DY, Stefanovic N et al (2010) Antiatherosclerotic and renoprotective effects of ebselen in the diabetic apolipoprotein E/GPx1-double knockout mouse. Diabetes 59:3198–3207
Tikellis C, Bialkowski K, Pete J et al (2008) ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes 57:1018–1025
Wang B, Jha JC, Hagiwara S et al (2014) Transforming growth factor-β1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int 85:352–361
Coughlan MT, Thallas-Bonke V, Pete J et al (2007) Combination therapy with the advanced glycation end product cross-link breaker, alagebrium, and angiotensin converting enzyme inhibitors in diabetes: synergy or redundancy? Endocrinology 148:886–895
Watson AM, Li J, Samijono D et al (2014) Quinapril treatment abolishes diabetes-associated atherosclerosis in RAGE/apolipoprotein E double knockout mice. Atherosclerosis 235:444–448
Candido R, Jandeleit-Dahm KA, Cao Z et al (2002) Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 106:246–253
Watson AM, Olukman M, Koulis C et al (2013) Urotensin II receptor antagonism confers vasoprotective effects in diabetes associated atherosclerosis: studies in humans and in a mouse model of diabetes. Diabetologia 56:1155–1165
Gray SP, Di Marco E, Kennedy K et al (2016) Reactive oxygen species can provide atheroprotection via nox4-dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol 36:295–307
Jha JC, Thallas-Bonke V, Banal C et al (2016) Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia 59:379–389
You YH, Quach T, Saito R, Pham J, Sharma K (2016) Metabolomics reveals a key role for fumarate in mediating the effects of NADPH oxidase 4 in diabetic kidney disease. J Am Soc Nephrol 27:466–481
Gorin Y, Cavaglieri RC, Khazim K et al (2015) Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am J Physiol Renal Physiol 308:F1276–F1287
Wilkinson-Berka JL, Deliyanti D, Rana I et al (2014) NADPH oxidase, NOX1, mediates vascular injury in ischemic retinopathy. Antioxid Redox Signal 20:2726–2740
Coughlan MT, Thorburn DR, Penfold SA et al (2009) RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol 20:742–752
Kuroda J, Ago T, Nishimura A et al (2014) Nox4 is a major source of superoxide production in human brain pericytes. J Vasc Res 51:429–438
Zheleznova N, O’Connor P, Cowley A (2015) Role of NOX4 in superoxide production stimulated by H+ efflux in mTAL of SS rat. The FASEB Journal 29:S963.10
Lu Q, Zhai Y, Cheng Q et al (2013) The Akt-FoxO3a-manganese superoxide dismutase pathway is involved in the regulation of oxidative stress in diabetic nephropathy. Exp Physiol 98:934–945
Genkyotex (2015) Genkyotex announces top-line results of phase 2 clinical program. Available from www.genkyotex.com/genkyotex/index.cfm/news-events/genkyotex-announces-top-line-results-of-phase-2-clinical-program/. Accessed 1 Sept 2015
Acknowledgements
The authors thank M. Arnstein and K. Gilbert for their technical assistance (Baker IDI Heart & Diabetes Institute, Melbourne, VIC, Australia). Some of the data reported were presented as an abstract at the 52nd EASD Annual Meeting in Munich in 2016.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Funding
This work was supported by the National Health & Medical Research Council (NHMRC) of Australia, the JDRF, the Diabetes Australia Research Trust, Genkyotex SA research funding and the FP7 framework programme. KAMJ-D is supported by an NHMRC Senior Research Fellowship and MEC is a Senior Principal Research Fellow for the NHMRC and a previous JDRF Scholar. SPG is supported by the Australian Diabetes Society Skip Martin Early Career Fellowship. HHHWS is supported by the EU (Marie Curie International Reintegration Grant and the European Research Council Advanced Grant 30983347E RADMED). This work was supported in part by the Victorian Government’s Operational Infrastructure Support Program. The study sponsor was not involved in the design of the study; the collection, analysis, and interpretation of data; writing the report; or the decision to submit the report for publication.
Duality of interest
KAMJ-D has served on a Genkyotex advisory panel and CS is a paid employee of and owns shares in Genkyotex, Geneva, Switzerland. HHHWS is a co-founder of and minor shareholder in Vasopharm, Würzburg, Germany. All other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Contribution statement
SPG, MEC, KAMJ-D and HHHWS designed experiments and wrote and revised the manuscript. SPG, JCJ, KK, EVB and PC performed the experiments and analysed data. JCJ, KK, EVB and PC provided input to drafting the manuscript. CS provided the GKT137831 compound, was consulted on study design and contributed to manuscript revision. RT was consulted on study design and manuscript preparation. All authors provided final approval of the manuscript prior to submission for publication. SPG and KAMJ-D are responsible for the integrity of the work as a whole.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 404 kb)
Rights and permissions
About this article

Cite this article
Gray, S.P., Jha, J.C., Kennedy, K. et al. Combined NOX1/4 inhibition with GKT137831 in mice provides dose-dependent reno- and atheroprotection even in established micro- and macrovascular disease. Diabetologia 60, 927–937 (2017). https://doi.org/10.1007/s00125-017-4215-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4215-5