



Article Figures & Data
Figures
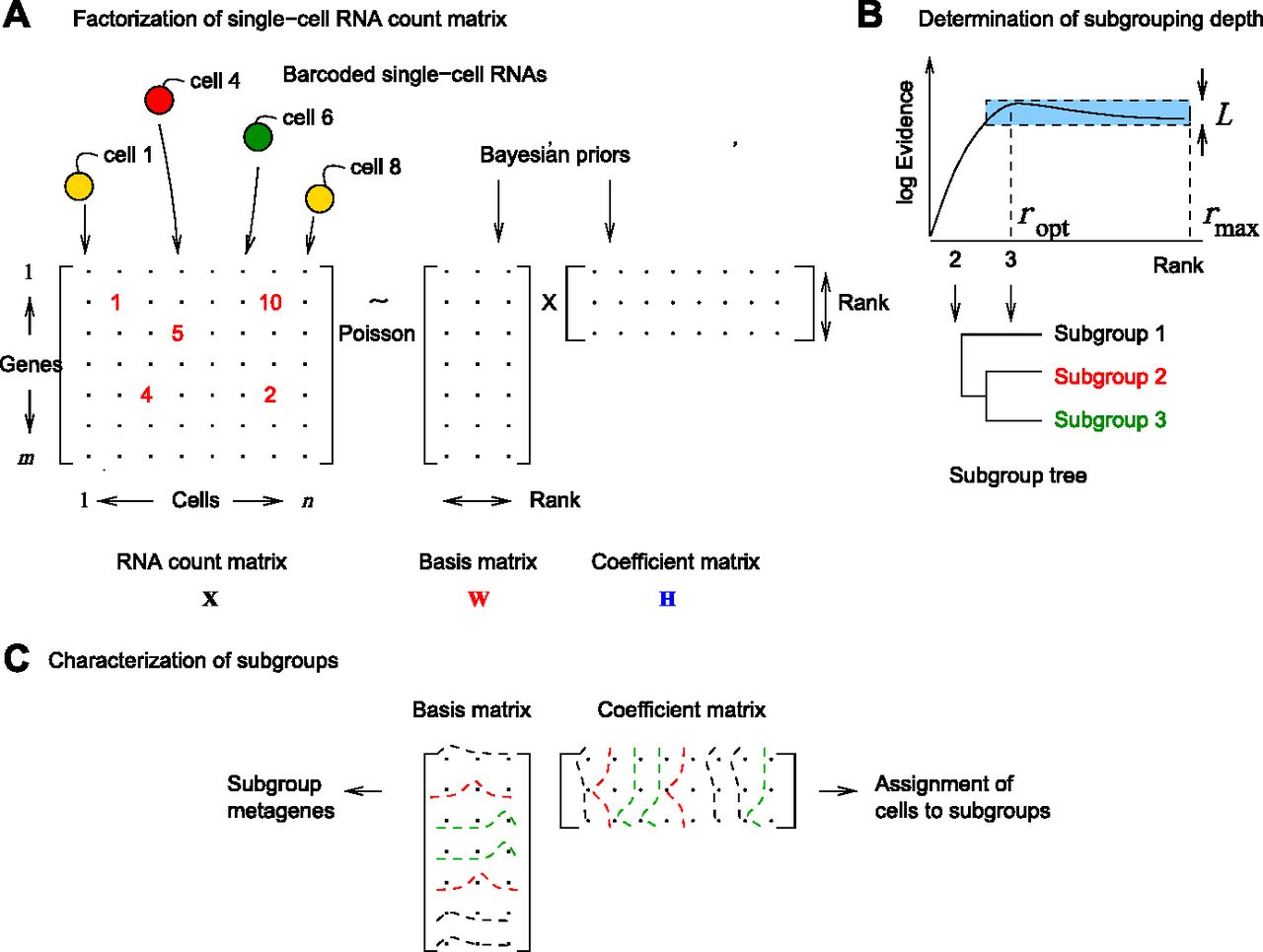
- Figure 1. bNMF for single-cell RNA-seq clustering.
(A) RNA count matrix derived from droplet-based single-cell RNA-seq data is modeled as a Poisson realization of the mean given by a product of basis W and coefficient H matrices sharing a common dimension rank. Factorization infers these matrices for varying rank values using gamma priors. (B) We find the optimal rank maximizing log evidence or marginal likelihood of hyperparameters given the data. Heterogeneity class is determined by the shape of evidence profile: in type I, the difference in evidence between the maximum at rank ropt and the value at rmax is larger than the threshold L; in type II, this difference is within L. The threshold is given by
- Figure 2. Comparison of optimal rank determination by NMF (ML-NMF and bNMF) and other clustering methods.
(A–D) Simulated data of 100 genes and 10 subgroups of cells (20 in each subgroup; 200 in total, except noted otherwise in (C)). ML-NMF narrows down the rank into an optimal range based on two quality measures, dispersion and cophenetic coefficient. (C) bNMF finds the correct rank 10 maximizing evidence. (D) Seurat (Macosko et al, 2015) requires specification of resolution parameter; the correct number of subgroups is reached as the upper bound with respect to resolution. (E, F) ML-NMF applied to PBMC single-cell data (Zheng et al, 2017). (G) bNMF applied to PBMC data sets of different sizes led to the optimal rank maximizing evidence as
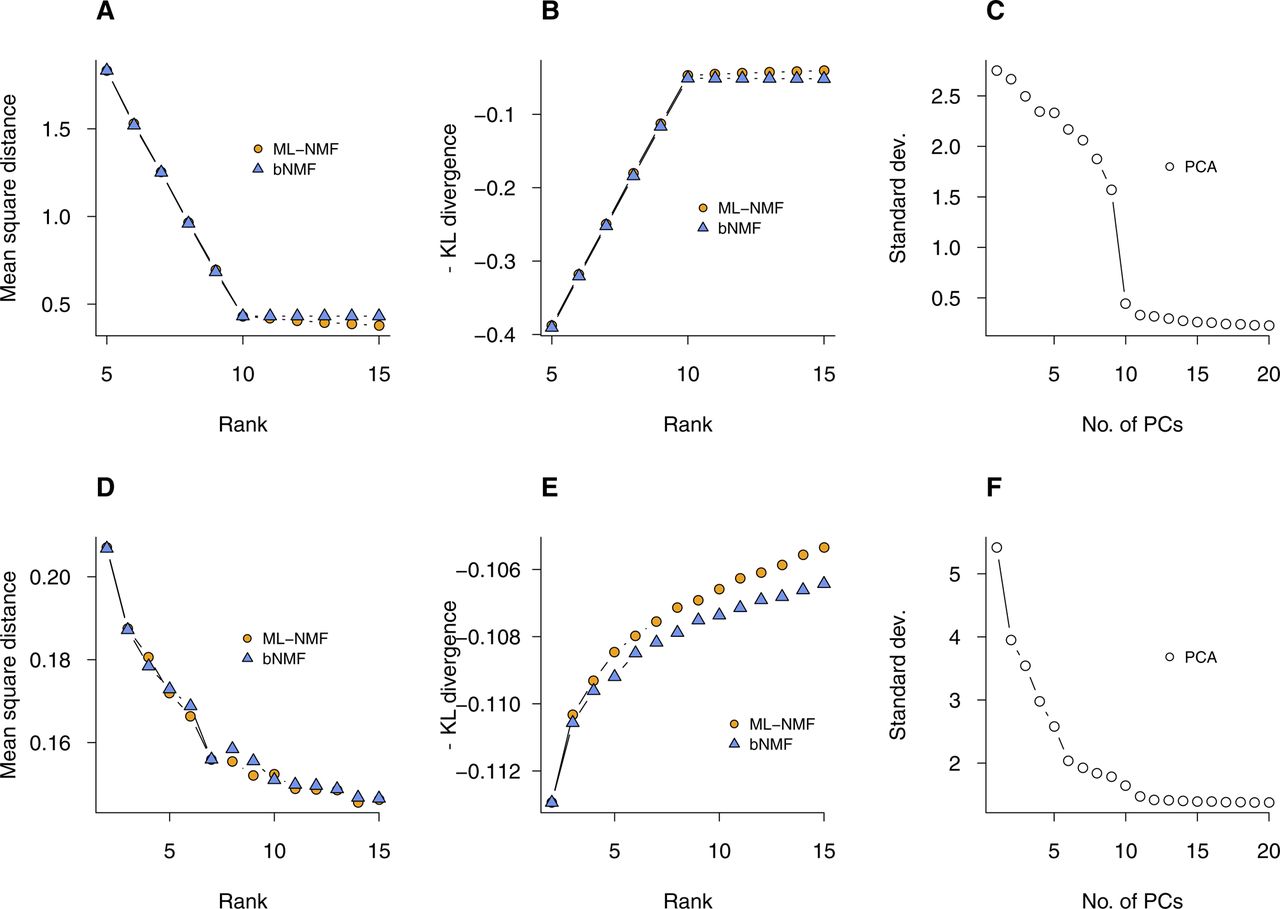
- Figure S1. Quality measures from ML-NMF and bNMF for simulated and PBMC data.
See Fig 2. (A–C) Euclidean distance, negative generalized Kullback–Leibler (KL) divergence, and SD from ML-NMF, bNMF, and PCA-based Seurat, applied to simulated data of Fig 2A–D. (D–F) Analogous results for PBMC data (Fig 2E–H).
- Figure S2. Outlier cells in bNMF factorization.
(A, B) A simulated data set generated as in Fig 2A–D was used with rank 5 and factorized using bNMF. (C) Outliers (white symbols) were identified using minimum covariance determinant method (Hubert & Debruyne, 2010).
- Figure S3. Convergence of bNMF algorithm tested with simulated data sets.
Two factor matrices W and H with dimensions 100 × 5 and 5 × n (n = 10, 100, and 1,000) were generated by sampling their elements from gamma priors
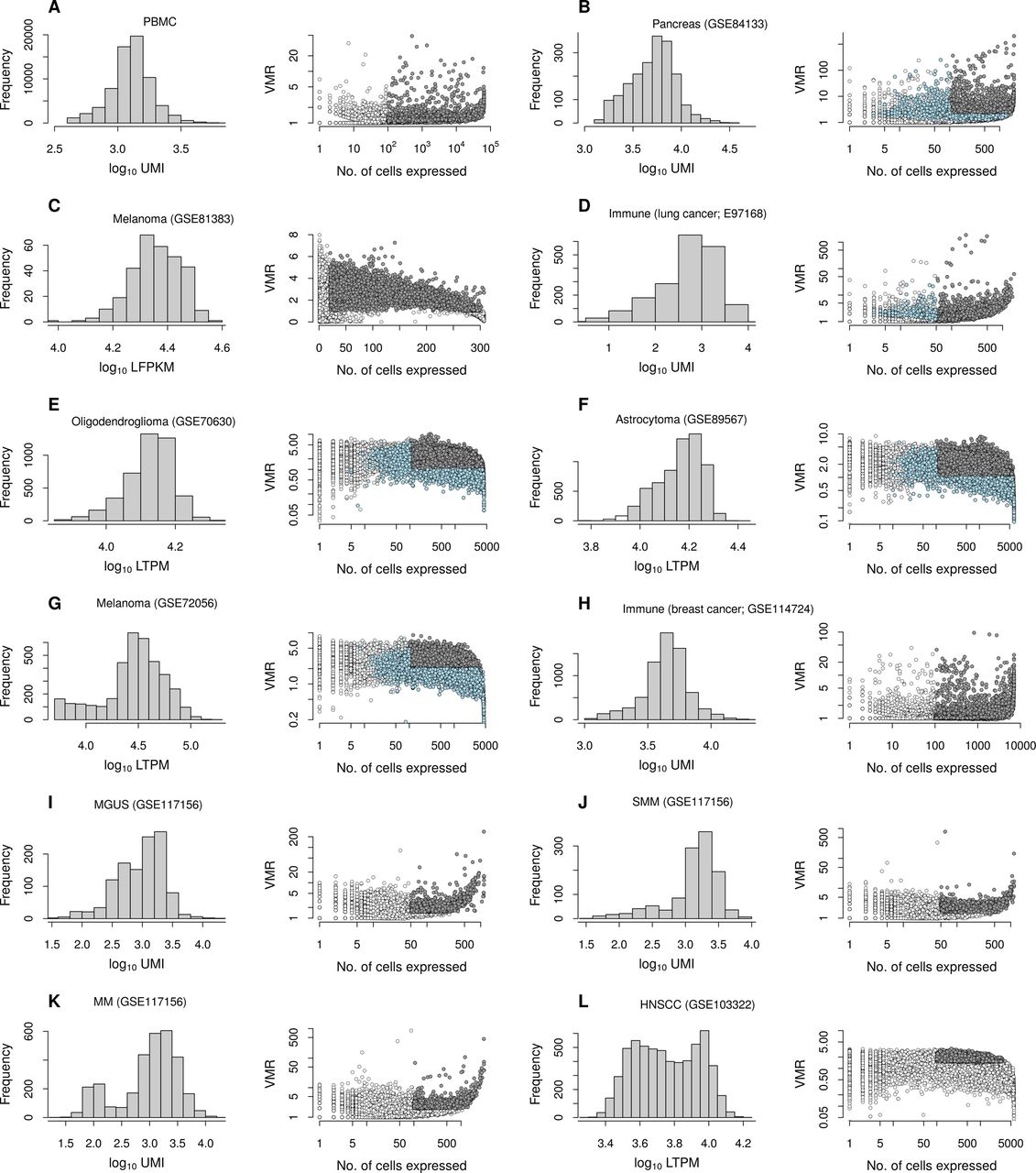
- Figure S4. Distributions of total RNA count and expression variances for data sets used.
(A–L) Total RNA counts for cells and VMR of genes are shown in the left and right columns, respectively. Filled circles are genes selected for downstream analysis, which include genes with VMR and number of cells expressing genes above thresholds (gray), and additional genes with modes in their count distribution (light blue), if included. See Table S1. LFPKM, log2 (1 + FPKM); FPKM, fragments per kilobase per million reads; LTPM, log2 (1 + TPM); TPM, transcripts per million reads.
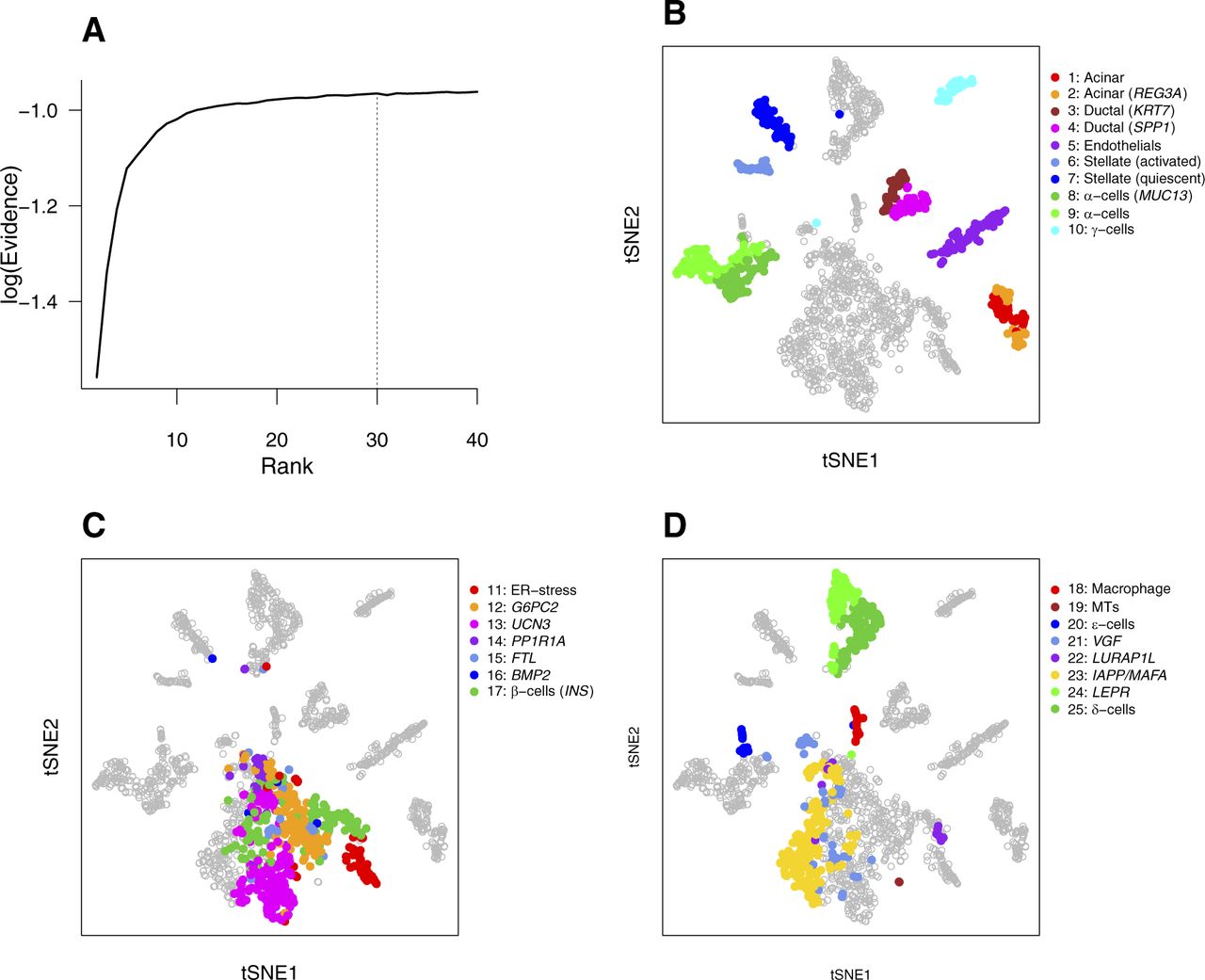
- Figure 3. bNMF subgrouping results for PBMC and pancreas data sets.
(A–C) Results for the PBMC data set (n = 34,289). (A) Metagenes for subgroups derived from the factor matrix W under optimal rank 9 (Fig 2G). Heat map shows the relative magnitudes of matrix element Wik for each gene i and subgroup k, rescaled such that in each row, minimum and maximum correspond to 0 and 1. Up to 10 metagenes in addition to preselected markers per subgroup are shown. (B) Subgroup tree showing hierarchical relationships between subgroups under varying ranks from the lowest (2) to the optimal (9). Branching of a subgroup under a given rank into two under a successively larger rank was inferred by applying the majority rule (see the Materials and Methods section). (C) Visualization of subgroups with tSNE. Subgroup ID and composition of cells are indicated. (D, E) Comparison of cell type compositions predictedby bNMF and bulk data deconvolution method, CIBERSORT (Newman et al, 2015). Outcomes for the full fresh PBMC data and an example mixture of seven purified cell types are shown in (D) and (E), respectively. (F) Subgrouping of human pancreas cell data (Baron et al, 2016). Colors indicate major cell types. Insulin-producing β-cells are in yellow (see Figs S5 and S6 and Table S2). MT, metallothionein. (G) Mean RNA count of INS gene in each pancreas subgroup.
- Figure S5. bNMF clustering outcomes of human pancreatic cells (Baron et al, 2016).
(A) Rank versus evidence profile. The heterogeneity class is type II and ropt = 30. (B–D) tSNE visualization of 25 subgroups. See Fig 3F and G.
- Figure S6. Top 20 metagenes of 25 subgroups derived from human pancreatic cell data (Baron et al, 2016).
Scores are magnitudes of coefficient matrix H elements rescaled by the geometric mean within each row corresponding to genes.
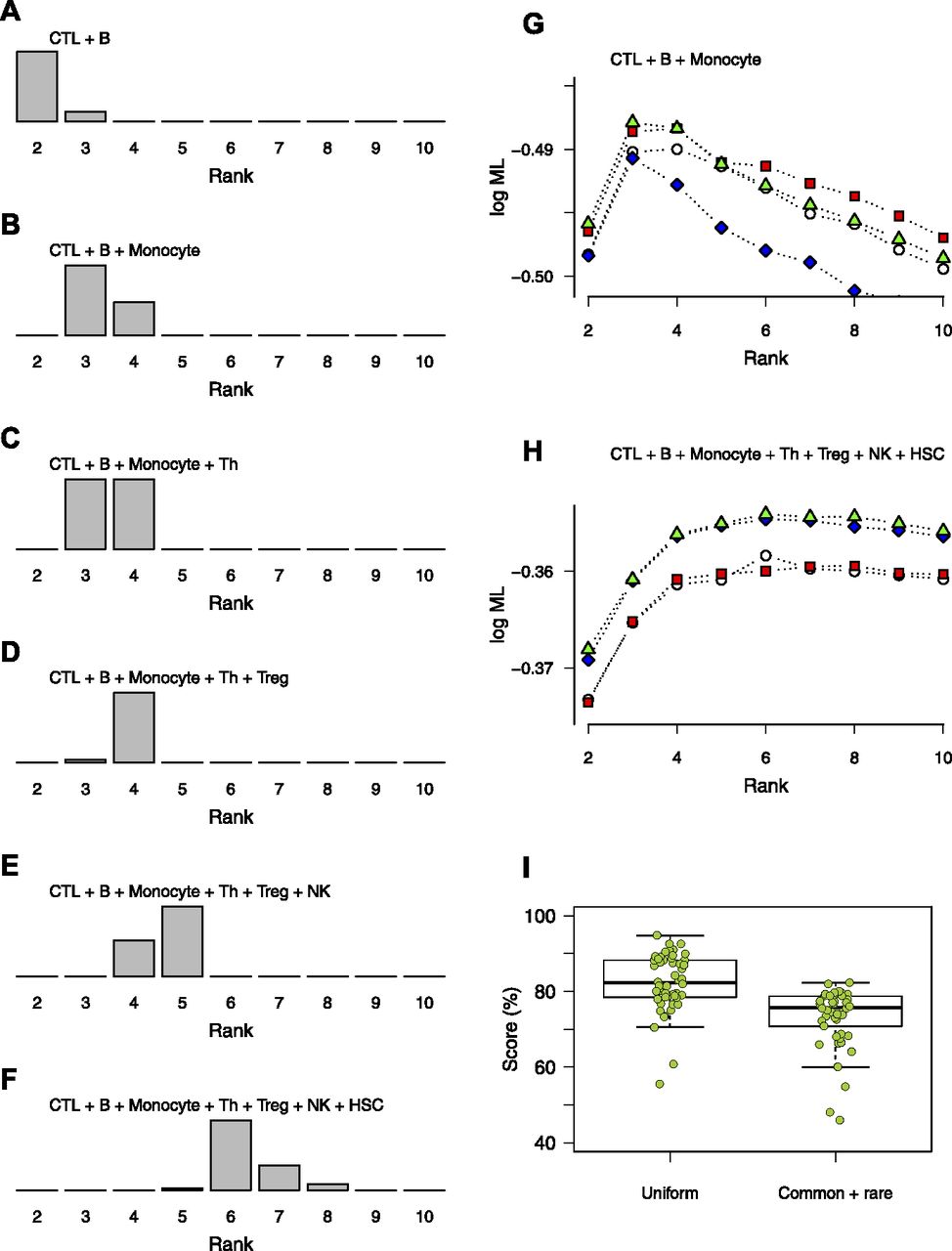
- Figure 4. Distributions of optimal ranks from bNMF inference applied to randomly sampled mixtures of purified blood cells.
(A–F) Mixtures containing selections of CD8+ T cells (CTL), B cells (B), monocytes, CD4+ T cells (Th), regulatory T cells (Treg), NK cells (NK), and CD34+ HSCs, of varying compositions as indicated. (G, H) Examples of rank versus evidence profiles for mixtures of three (G) and seven (H) blood cell types. (I) Subgroup assignment scores (fraction of correctly assigned cells) of bNMF-based inferences applied to mixtures of four purified blood cell types shown in (C). Two sets of mixtures with different compositions were sampled, one with uniform cell counts (“uniform”) and the other where three cell types were ∼10% in count than the rest (“common + rare”). Mean scores are 0.82 (0.08, SD) and 0.73 (0.08) for uniform and common + rare cases, respectively.
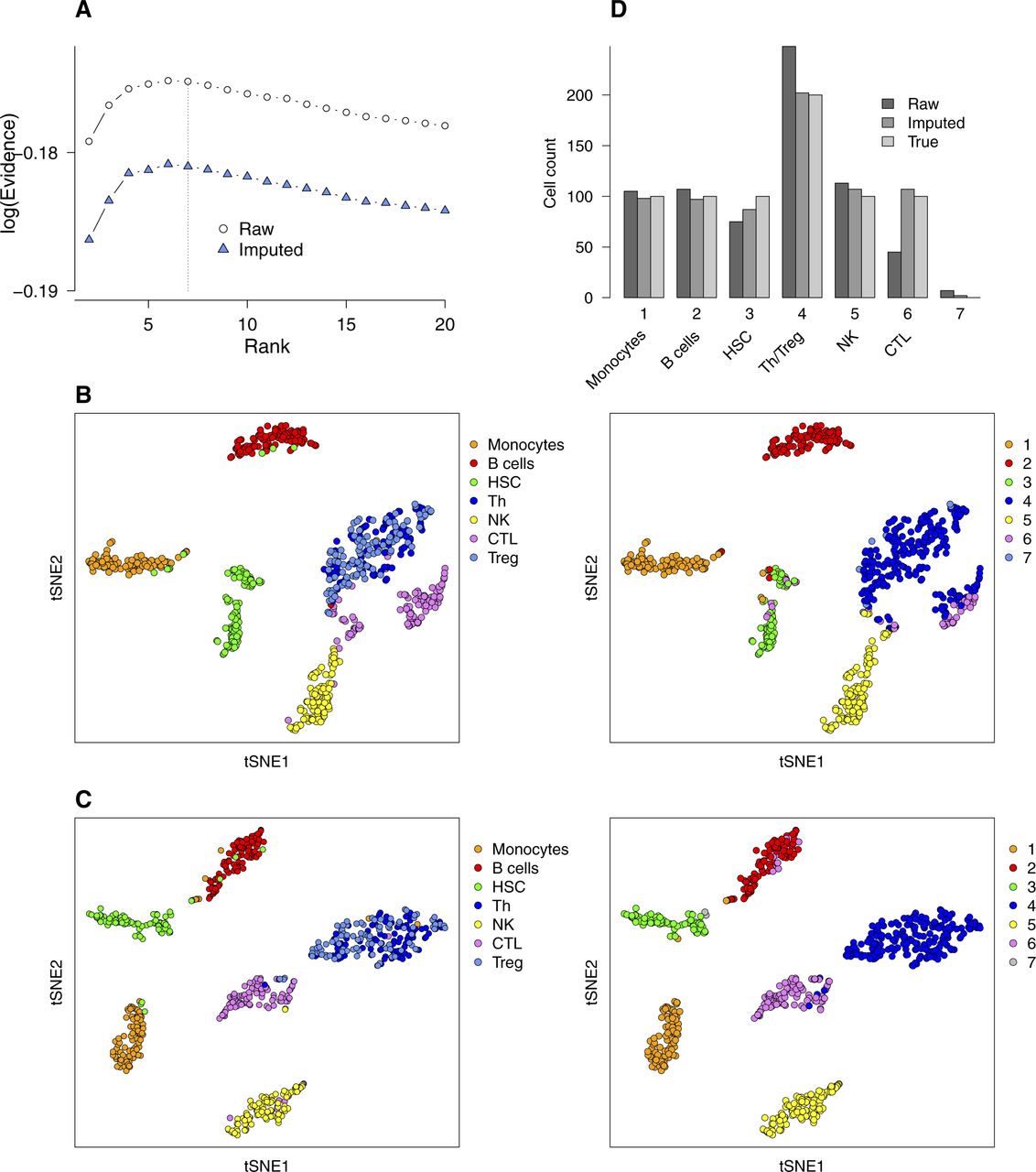
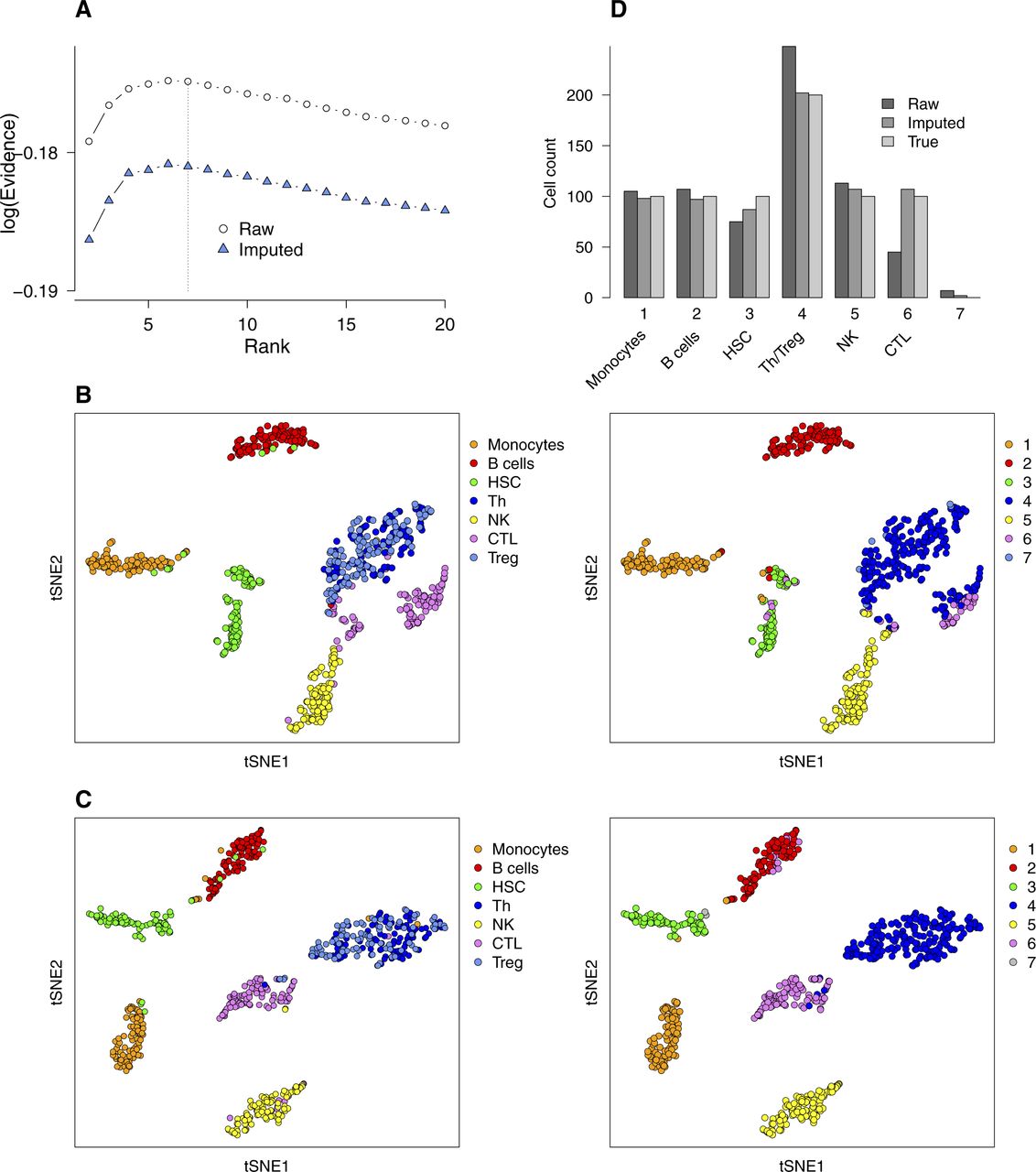
- Figure S7. Comparison of bNMF clustering applied to a PBMC mixture without and with imputation of RNA counts.
A mixture of seven immune cell types (monocytes, B cells, CD34+ HSCs, helper T cells [Th], natural killer [NK] cells, cytotoxic T lymphocytes [CTL], and Treg; n = 100 each; see Fig 4F) was used. (A) Evidence profiles. (B, C) tSNE maps (derived from raw count matrix, (B); derived from imputed count matrix, (C); colored by cell types of origin [left column] and by cluster assignment from H matrix [right column; rank 7]). Note that Th and Treg cells are grouped together in cluster 4. (D) Cell counts in each cluster.
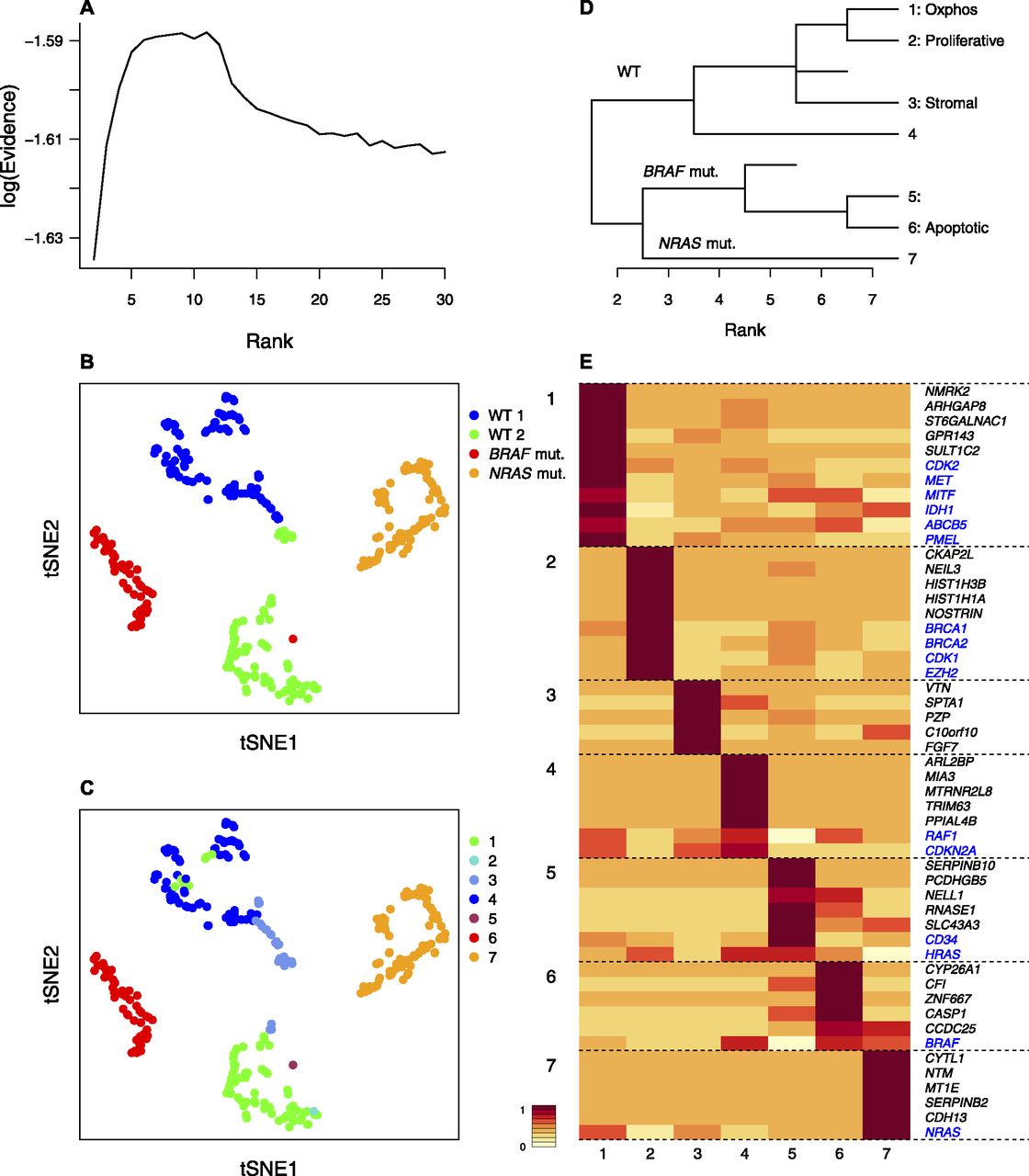
- Figure 5. bNMF subgrouping results of melanoma cell culture transcriptome data (Gerber et al, 2017).
(A) Rank versus evidence profile. (B, C) tSNE visualizations of cells using bNMF H matrix elements colored by sample of origin in (B) and subgroup identity under rank 7 in (C). (D) Custer tree from rank 2 to 7. Oxphos, oxidative phosphorylation (Gerber et al, 2017). (E) Metagene map showing top five metagenes in each subgroup and marker genes (blue). mut., mutant.
- Figure 6. bNMF clustering outcomes for in vivo solid tumor and myeloma samples.
(A–F) Rank versus evidence profiles of data sets with accession numbers as indicated (Tirosh et al, 2016a, 2016b; Lavin et al, 2017; Puram et al, 2017; Venteicher et al, 2017; Azizi et al, 2018). Dotted blue lines are smooth-spline fits to data. Vertical dashed lines are locations of optimal rank. (G–I) Evidence profiles of MM samples in MGUS, SMM, and full MM stages (Ledergor et al, 2018).
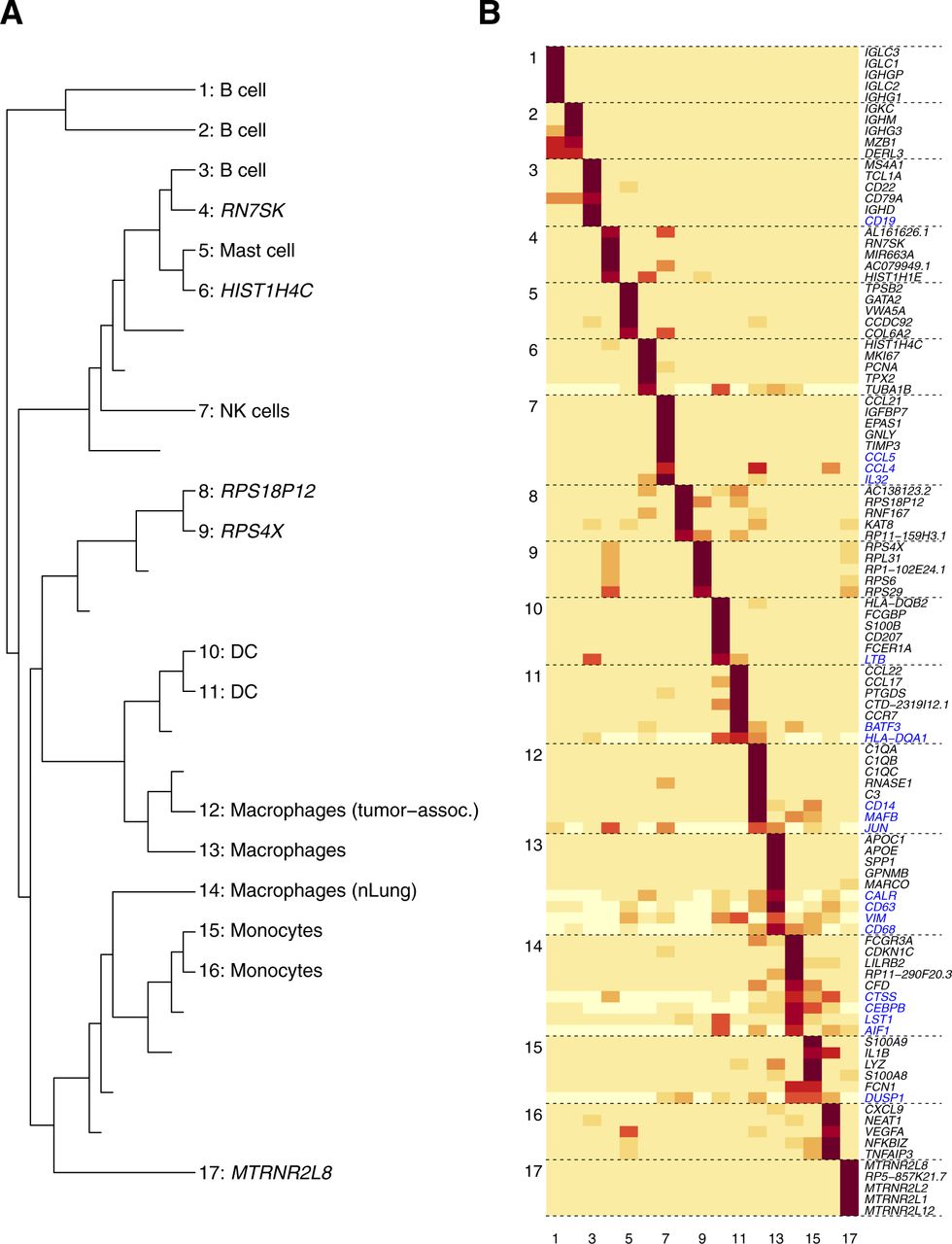
- Figure S8. bNMF clustering outcome for innate immune cell data from lung cancer patients.
(A) Subgroup tree of connecting r = 2–17 annotated with cell types assigned by marker genes from Lavin et al (2017). (B) Metagene map for 17 subgroups. Genes labeled in blue are marker genes ranked below five.
- Figure S10. bNMF subgroups in head and neck cancer data set (Puram et al, 2017).
(A) Subgroup tree. (B) Metagene map (top five plus marker genes in blue) for 15 subgroups. (C) tSNE visualization of 15 subgroups. Numbers indicate subgroup labels in (A–B). (D) Cell-type annotation by Puram et al (2017).
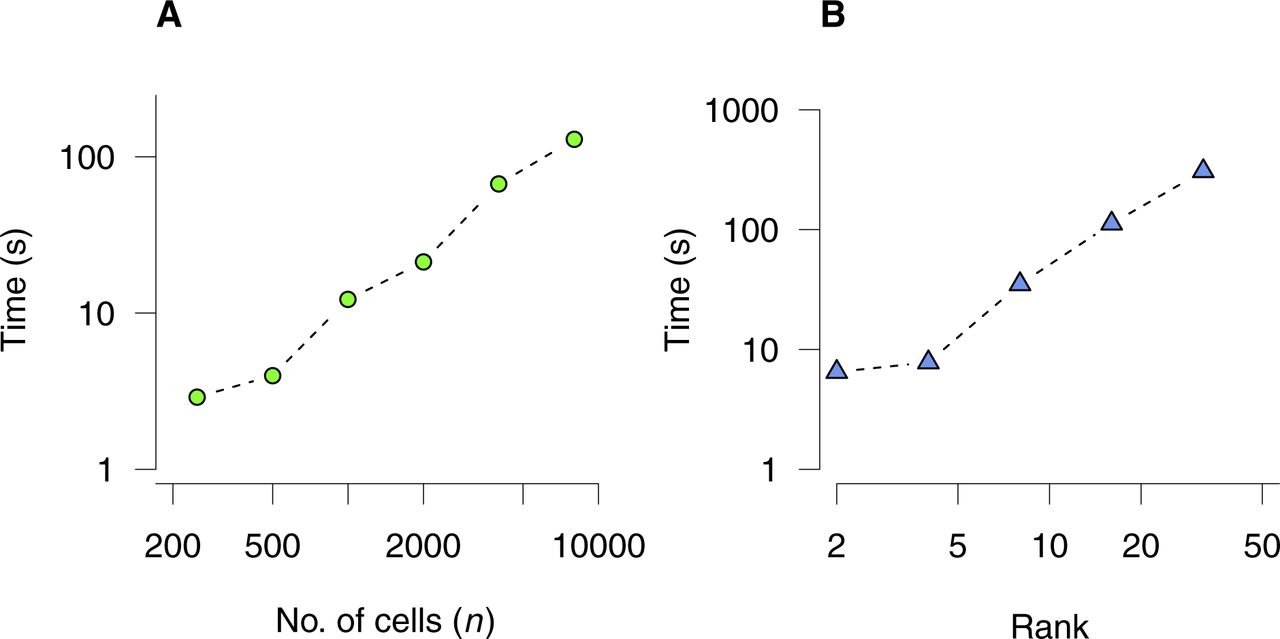
- Figure S11. Computational load for bNMF inference.
Execution time is shown for factorizing a count matrix using bNMF under a single initial condition until convergence. (A) Matrices with m = 500 rows under rank r = 5 with varying number of cells. (B) Matrices with m = 500 rows and n = 1,000 cells under varying rank values. An Intel Core i5 CPU with 2.5 GHz was used.











Supplementary Materials




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
