Article Text

Abstract
Baller-Gerold syndrome (BGS) is a rare autosomal recessive condition with radial aplasia/hypoplasia and craniosynostosis (OMIM 218600). Of >20 cases reported so far, a few appear atypical and have been reassigned to other nosologic entities, including Fanconi anaemia, Roberts SC phocomelia, and Pfeiffer syndromes after demonstration of corresponding cytogenetic or molecular abnormalities. Clinical overlap between BGS, Rothmund-Thomson syndrome (RTS), and RAPADILINO syndrome is noticeable. Because patients with RAPADILINO syndrome and a subset of patients with RTS have RECQL4 mutations, we reassessed two previously reported BGS families and found causal mutations in RECQL4 in both. In the first family, four affected offspring had craniosynostosis and radial defect and one of them developed poikiloderma. In this family, compound heterozygosity for a R1021W missense mutation and a g.2886delT frameshift mutation of exon 9 was found. In the second family, the affected male had craniosynostosis, radial ray defect, poikiloderma, and short stature. He had a homozygous splice site mutation (IVS17-2A>C). In both families, the affected offspring had craniosynostosis, radial defects, and growth retardation, and two developed poikiloderma. Our results confirm that BGS in a subgroup of patients is due to RECQL4 mutations and could be integrated into a clinical spectrum that encompasses RTS and RAPADILINO syndrome.
- BGS, Baller-Gerold syndrome
- OFC, occipito-frontal circumference
- RTS, Rothmund-Thomson syndrome
- craniosynostosis
- limb defect
- poikiloderma
- radial ray
- skin
Statistics from Altmetric.com
Bilateral radial ray hypoplasia is found in a number of multiple malformation syndromes such as Fanconi anaemia (OMIM 227650), Roberts SC phocomelia (OMIM 269000), thrombocytopenia-absent radius syndrome (OMIM 274000), Holt-Oram syndrome (OMIM 142900), and SALL4 related syndromes.1 Craniosynostosis, when symmetrical and involving coronal and lambdoid sutures, may be indicative of at least 50 syndromes.2 However, these two distinctive clinical features are associated in a limited number of syndromes, most constantly in Baller-Gerold syndrome (BGS; OMIM 218600). BGS was delineated after Baller3 and Gerold4 described the first patients with radial hypoplasia associated with craniosynostosis. Interestingly, the first patient has (as far as can be discerned from the poor quality clinical photograph of the original paper) a skin appearance that seems compatible with poikiloderma (fig 1).3

Clinical photograph of Baller’s original patient retrieved from the German literature.3 Note brachycephaly and the skin appearance of her forearm.
A limited number of additional patients have been reported since then.5,6,7,8,9,10,11,12 Clinical overlap with other syndromes became more obvious when clinical diagnoses of BGS were subsequently challenged by cytogenetic or molecular tests that revealed a diagnosis of Fanconi anaemia,13–18 Roberts SC syndrome,19 or Saethre-Chotzen (OMIM 101400) syndrome. TWIST mutations are found in the latter condition and are usually associated with a broad fingers-receding forehead craniosynostosis phenotype, but anecdotal patients may also display radial hypoplasia.20,21 The features of some patients with FGFR2 mutations may also mimic those of BGS but to a lesser extent, as exemplified by a patient who also has humero-ulnar synostosis.22 The same holds true for SALL4 related syndromes.1 Given the phenotypic overlap between these different conditions, additional studies, such as chromosome breakage assays or DNA sequencing of the FGFR or TWIST genes, may be useful to exclude other syndromes depending on the clinical findings. We hereby update reports of two previously published BGS families11,12 and correlate the findings with the identification of mutations in the RECQL4 gene including two novel mutations, g.5428A>C and g.5435C>T.
Clinical histories
Family 1
Patient 1
The index patient has been described previously.11 He was born at term after an uneventful pregnancy to unrelated parents. He had severe radial ray hypoplasia with oligodactyly, anus anteposition, pes talus, multiple cranial suture synostosis, and a distinctive facial dysmorphia with a very small mouth, thin vermilion border, long upper lip, and microretrognathism (fig 2, middle panel). He died of unknown cause a few minutes after birth.

Family 1: Note the clinical findings in a 16 week old fetus (patient 3) displaying mild brachycephaly, radial aplasia, and oligodactyly (left panel). These findings have to be compared with the newborn infant (patient 1) who has marked brachycephaly and facial dysmorphia that includes a small mouth, short nose, short palpebral fissures with telecanthus, and a bulging forehead where a W-shaped upper furrow points to a widely opened anterior fontanelle (middle panel). At an older age (patient 4 at 3 and 6 years), dysmorphia is less pronounced but failure to thrive is obvious. On a 3D tomodensitometric reconstruction of the skull, one can see the sharp contrast between coronal and lambdoid craniosynostosis and skull ossification defect with wide fontanelles (right panel). (Written authorisation has been obtained from the legal tutors for the publication of these clinical photographs.)
Patients 2 and 3
Subsequently, the parents had two other affected fetuses. Upper limb shortening, oligodactyly, and severe brachycephaly were detected at ultrasound screening at 24 and 16 weeks, respectively. Both pregnancies were terminated. In the first case (24 weeks), necropsy demonstrated a male with normal weight (470 g), craniosynostosis of lambdoid and coronal sutures, a large anterior fontanelle, and radial ray hypoplasia with thumb aplasia (fig 2, left panel). Necropsy of the second fetus at 16 weeks, a female weighing 182 g, was similar in many respects and demonstrated turricephaly, coronal craniosynostosis, large metopic and frontal sutures, and wide fontanelles with a normal brain. Arms were short and bowed. There was also radial aplasia, anus anteposition, and hypoplasia of the great toe.
Patient 4
After the birth of a normal boy, a fifth pregnancy was also carefully monitored by ultrasound. Upper limbs shortening and bilateral thumb agenesis were diagnosed at 15 weeks. The couple decided to continue the pregnancy. Patient 4 was born at term (fig 2, right panel). She weighed 2680 g at birth and was 46 cm long, and her occipito-frontal circumference (OFC) was 36 cm. She had acrocephaly, temporal bulging, a widely open anterior fontanelle, midface retraction with saddle nose, and bilateral agenesis of the thumbs. On x rays, ulna and radius were normally shaped. Anus was anteposed. Surgical correction of the craniosynostosis and pollicisation of the right index were successful. Patellar hypoplasia was noted at the age of 6 months. Erythematous skin lesions appeared progressively on the face and limbs during the first months of life, leading to a diagnosis of poikiloderma. Skin atrophy became more obvious over time but kept the same topographic pattern: almost absent on the trunk, mild on thighs, moderate on cheeks, nose and forehead, and most prominent on forearms and hands, with the intensity of the erythema fluctuating depending on sun exposure. Growth rate was abnormal from birth, leading to progressive dwarfism. At the age of 9 months, she was 64 cm tall (−1.7 SD) and weighed 5500 g (−3.2 SD). At the age of 3 years, she was 79 cm tall (−4.3 SD). At the age of 6, she was 93.3 cm tall (−4.4 SD) and weighed 11 kg (−3.2 SD), and her OFC was 48 cm (−3 SD). Endocrine work up included thyroid function and GH insulin tests, as well as somatomedin C and IGF1 plasma concentrations which were all normal. She had chronic feeding difficulties requiring nasal tube feeding during the first months of life. Low appetite persisted during childhood.
Early psychomotor development was normal. She walked unsupported at the age of 18 months. Absence of patellae induced an inward rotated legs’ gait. Major voice problems related to her short, hypomobile velum were not improved by speech therapy. Despite her marked expressive problems, she participated in a regular classroom at the primary school level.
Family 2
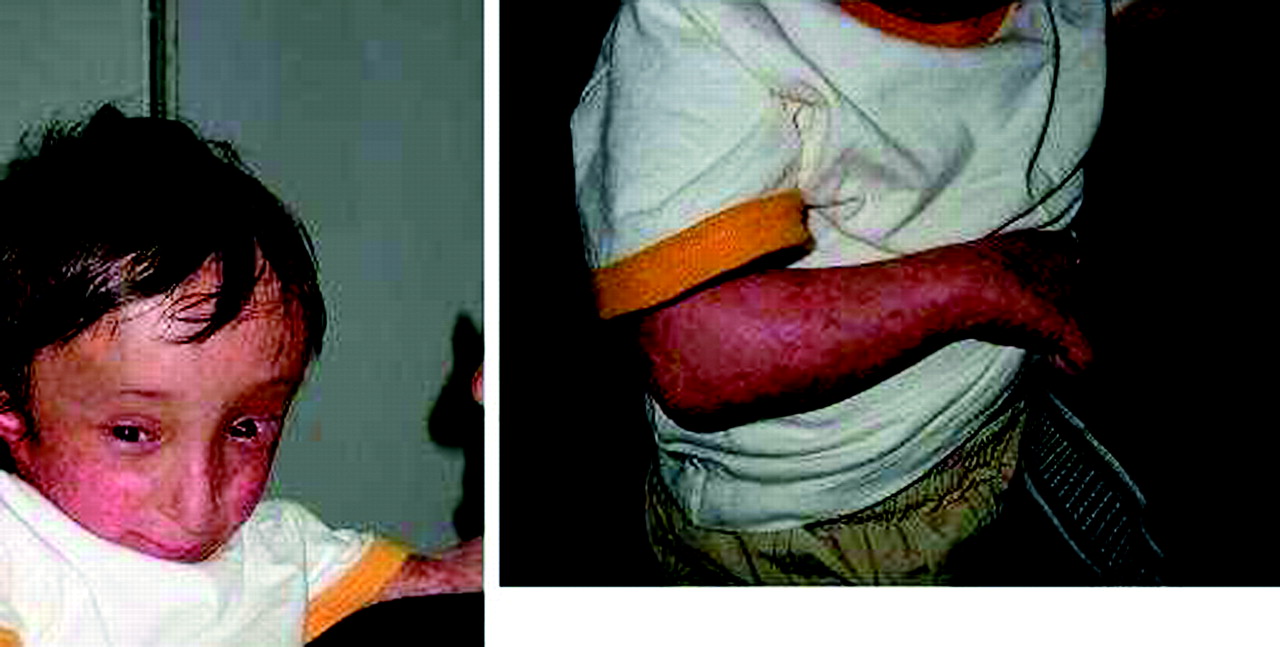
This boy has been described previously12 (fig 3). He was the second child of first cousins. The pregnancy was marked by severe intrauterine growth retardation. Clinical examination showed turribrachycephaly in relation to craniosynostosis of lambdoid and coronal sutures, short forearms with radial deviation of the hands, missing left thumb and rudimentary thumb on the right, short stature, and poikiloderma.
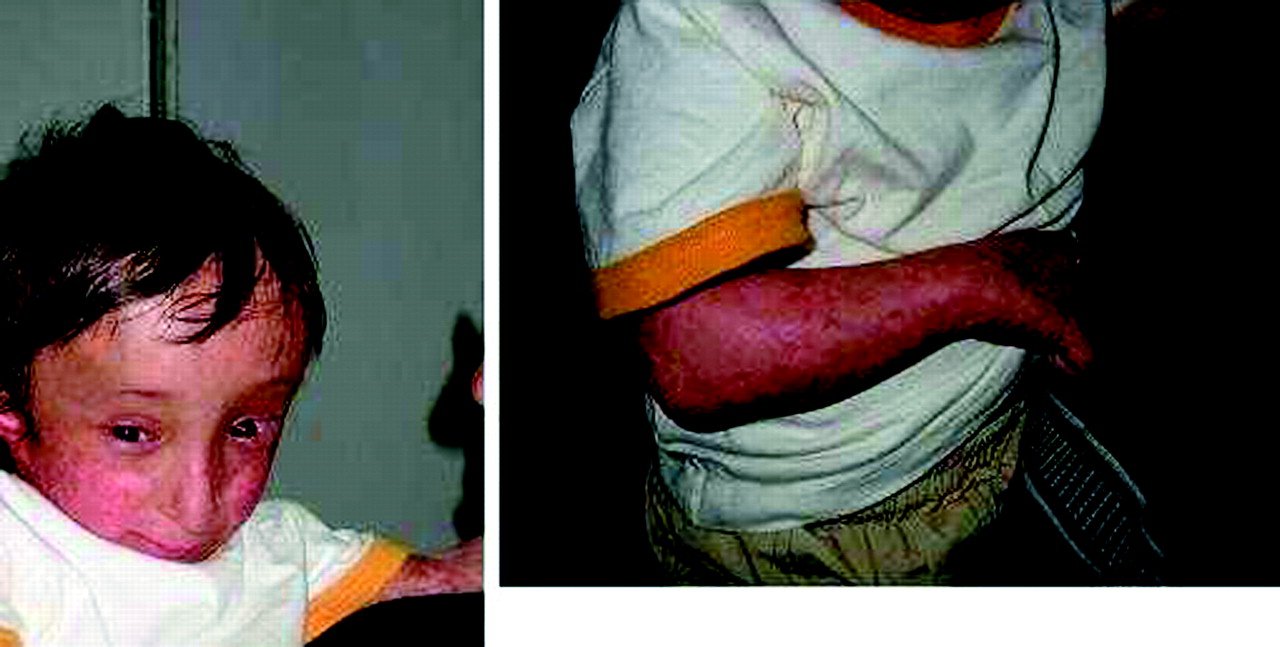
Index patient from family 2. Note similarities with patient 4 of family 1. (Written authorisation has been obtained from the legal tutors for the publication of these clinical photographs.)
Molecular studies
Molecular studies were performed on DNA extracted from peripheral leucocytes. Sequencing of FGFR2 exons IIIa and c, FGFR3 exon IIIa, and the TWIST gene was performed according to previously reported methods23 and was normal in the index patient of family 1. All exons and the short introns of RECQL4, except for a small part of intron 12, were also sequenced, including the consensus splice sites, in both families.24
In family 1, samples from parents and children 1, 4, and 5 were available. Samples from the first and fifth children revealed compound heterozygosity for two mutations: a substitution and deletion mutation (g.2881G>C; g.2886delT) in exon 9 (also found in the maternal allele) and a missense mutation (g.5435C>T/R1021W) in exon 18 (also found in the paternal allele). The g.5435C>T mutation causes the arginine to tryptophan amino acid substitution R1021W. The unaffected brother was found to have only the g.2881G>C; g.2886delT mutation. The g.2881G>C/S523T change is likely a polymorphism which cosegregates with the g.2886delT mutation.25,26 No other pathogenic mutation was detected in the affected children.
In family 2, samples from the parents and affected male were available. The patient was homozygous for a g.5428A>C mutation which changes the splice acceptor site IVS17-2A>C, probably affecting the correct splicing. Unfortunately RNA was not available to confirm the effect of the mutation at the transcript level.
The localisation of these BGS mutations is presented in fig 4 together with the reported RECQL4 mutations causing Rothmund-Thomson syndrome (RTS) or RAPADILINO syndrome.
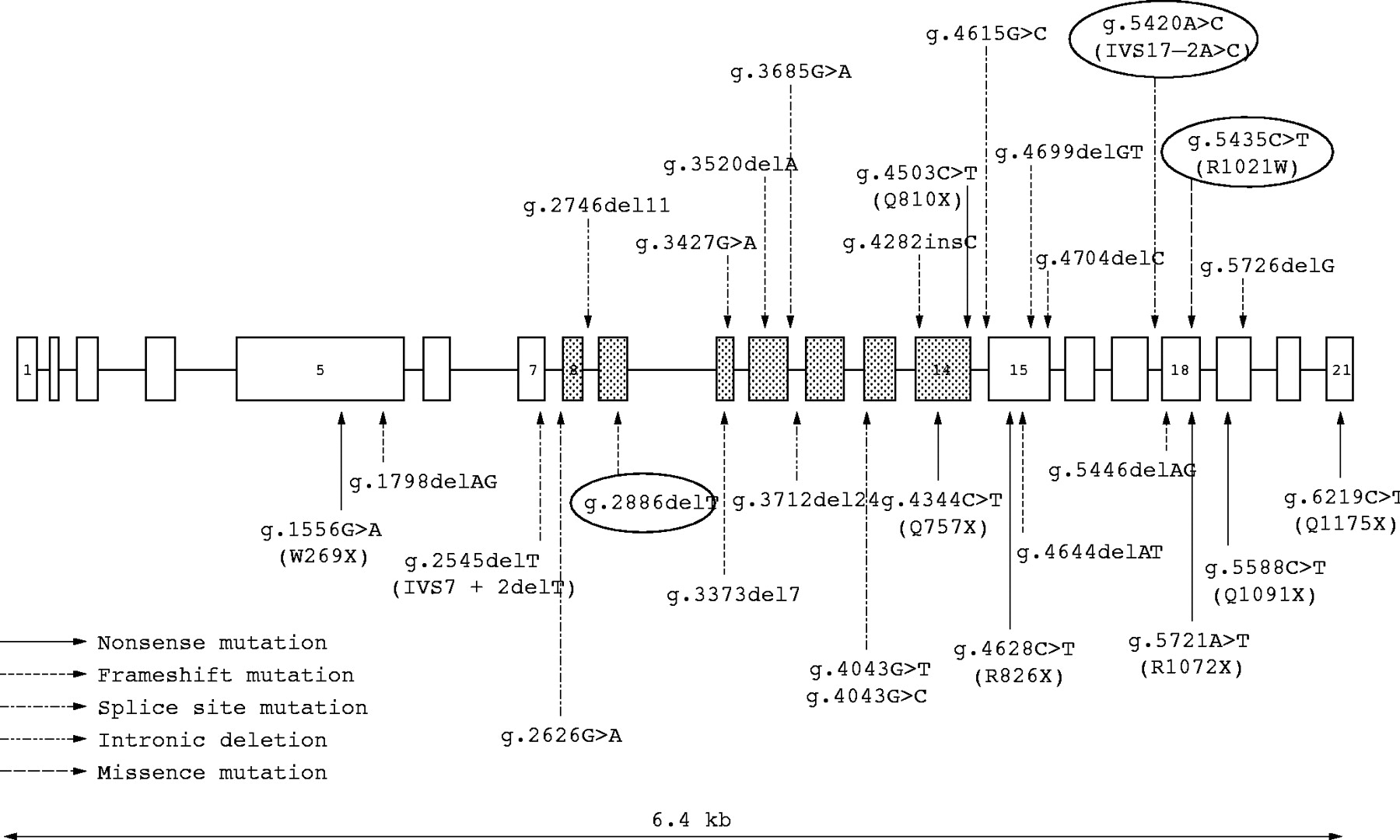
Schematic structure of the RECQL4 gene. Note the three new mutations (ringed) and compare them with the previously reported mutations in RTS (normal characters) and RAPADILINO syndrome (in bold).
DISCUSSION
When reviewing the published cases of BGS, the impression is that there is a core diagnosis consisting of lambdoid and coronal craniosynostosis in association with radial hypoplasia. The anecdotal reassignments of BGS published cases to other nosological entities were prompted by secondary haematological complications or atypical findings. Guided by clinical findings, one may therefore in specific circumstances, in addition to initial work up, require special chromosomal analysis after incubation with clastogens, or studies of DNA crosslinking sensitivity. Sequencing of FGFR1, FGFR2, FGFR3, or TWIST genes may also be required according to clinical findings.
Of note is the fact that poikiloderma is a skin manifestation that in RTS occurs after an interval of a few months. Caution should therefore be applied to BGS diagnoses made in the first few months of life and follow up evaluation is warranted.
Rothmund-Thomson syndrome (RTS; OMIM 268400) and RAPADILINO syndrome (OMIM 266280) are two recessively inherited syndromes displaying some clinical overlap with BGS (fig 5). RTS is characterised by poikiloderma congenita (usually first manifested between 4 and 6 months of age), alopecia, skeletal defects, dystrophic nails, abnormal teeth, cataracts, and small stature. Photosensitivity is highly variable. Radial ray hypoplasia or absent thumbs occur in a minority of cases. The clinical diagnosis rests on the poikilodermatous rash. If the onset or distribution of poikiloderma is atypical, then two additional features such as bone abnormalities, cataracts, or osteosarcoma are required for a diagnosis of probable RTS.27 Analysis of 33 RTS patients suggests that approximately 60% of patients with definite or probable RTS carry mutations in RECQL428 with the remainder of RTS due to mutations in gene(s) that have not yet been identified. RAPADILINO is a rare autosomal recessive malformation syndrome.29 The acronym refers to the main clinical features (radial ray defect; patellae hypoplasia or aplasia and cleft or highly arched palate; diarrhea and dislocated joints; little size and limb malformation; nose slender and normal intelligence). Fourteen patients have been diagnosed in Finland and only three in other countries.24,29–32 One of the non-Finnish patients was later rediagnosed as having RTS.33 It has recently been shown that RAPADILINO syndrome is caused by mutations in RECQL4.24

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Venn diagram of the main features of the three entities. If we hypothesise a continuum between them, then the core (overlapping) criteria would include growth deficiency, facial dysmorphia, and GI disturbance.
RECQL4 encodes a member of the RecQ helicase family based on sequence conservation.34 However, few functional studies have been performed. Recently, it has been reported that the RECQL4 proteins interact in the cytoplasm with ubiquitin ligases UBR1 and UBR2 proteins of the N-end rule pathway.35 The physiological significance of this interaction is unknown. The Fin-major mutation24 is a splice site mutation causing in-frame skipping of exon 7. All the Finnish patients are either homozygotes or heterozygotes for this mutation (fig 3).
Similarities between RTS and BGS have already been pointed out by one of us.12 RAPADILINO syndrome also fits into the BGS clinical spectrum since radial hypoplasia is one of the hallmarks of RAPADILINO syndrome, and one non-Finnish case was rediagnosed as RTS after the patient developed a poikiloderma-like rash at the age of 21 months. An intermediate phenotype with craniosynostosis, poikiloderma, and anteriorly placed anus has also been reported recently.36 The fact that the clinical course of patient 4 from family 1 and the index patient from family 2 had poikiloderma prompted us to investigate a possible continuum between these apparently distinct entities by searching for mutations in the RECQL4 gene. In both families, mutations in RECQL4 were found to be the cause of the BGS phenotype.
In conclusion, we have demonstrated in two unrelated families that RECQL4 mutations cause BGS. These results bring the number of clinical syndromes attributable to RECQL4 mutations to three: RTS, RAPADILINO, and BGS. The genotype-phenotype correlations in these syndromes need to be studied further. It will be interesting to see whether certain mutations always lead to distinct phenotypes or if the correlation is more complex. Since a BGS phenotype has already been associated with Fanconi anaemia and Roberts SC phocomelia, and with TWIST and FGFR2 mutations, careful clinical delineation will assist in defining this nosological entity. Radial defects are a variable feature associated with otherwise classical craniosynostosis gene mutations. This feature also belongs to cytogenetically defined disorders, such as Fanconi anaemia or Roberts SC phocomelia, and to some cases of VATER Association. In these two subsets, cutaneous manifestations are found only in Fanconi anaemia patients in the form of pigmentary changes. We here provide evidence that a third subgroup of BGS patients have a RECQL4 related phenotype with eventual developmental skin lesions which are not present at birth. In this context, the consistent presence of sutural anomalies in 4/4 affected patients from family 1 may be a mutation specific manifestation perhaps related to the specific domain of the protein altered by the missense mutation. Study of intrafamilal variability and genotype-phenotype correlations within the RECQL4 spectrum, based on a larger set of patients, will be necessary to determine whether the apparently distinctive phenotypes breed true and are linked to distinct mutations. Also, multiple malformation syndromes that include craniosynostosis and/or radial ray aplasia and/or poikiloderma should be investigated for RECQL4 mutations.
REFERENCES
Footnotes
-
Published Online First 17 June 2005
-
Competing interests: none declared
-
Consent has been given for the publication of the details in this report