Article Text

Abstract
Background Administration of granulocyte-macrophage colony stimulating factor (GM-CSF) relieves symptoms in Crohn's disease (CD). It has been reported that reduced GM-CSF bioactivity is associated with more aggressive ileal behaviour and that GM-CSF-null mice exhibit ileal barrier dysfunction and develop a transmural ileitis following exposure to non-steroidal anti-inflammatory drugs (NSAIDs). STAT5 signalling is central to GM-CSF action. It was therefore hypothesised that GM-CSF signalling in non-haematopoietic cells is required for ileal homeostasis.
Methods Bone marrow (BM) chimeras were generated by reconstituting irradiated GM-CSF receptor (gm-csfr) β chain or GM-CSF (gm-csf) deficient mice with wild type BM (WTBM→GMRKO and WTBM→GMKO). Intestinal barrier function and the response to NSAID-induced ileal injury were examined. Expression of gm-csf, gm-csfr or stat5 in Caco-2 and HT-29 intestinal epithelial cell (IEC) lines was knocked down and the effect of GM-CSF signalling on IEC survival and proliferation was determined.
Results Elevated levels of GM-CSF autoantibodies in ileal CD were found to be associated with dysregulation of IEC survival and proliferation. GM-CSF receptor-deficient mice and WTBM→GMRKO chimeras exhibited ileal hyperpermeability. NSAID exposure induced a transmural ileitis in GM-CSF receptor-deficient mice and WTBM→GMRKO chimeras. Transplantation of wild type BM into GM-CSF-deficient mice prevented NSAID ileal injury and restored ileal barrier function. Ileal crypt IEC proliferation was reduced in WTBM→GMRKO chimeras, while STAT5 activation in ileal IEC following NSAID exposure was abrogated in WTBM→GMRKO chimeras. Following knock down of gm-csf, gm-csfr α or β chain or stat5a/b expression in Caco-2 cells, basal proliferation was suppressed. GM-CSF normalised proliferation of Caco-2 cells exposed to NSAID, which was blocked by stat5a/b RNA interference.
Conclusions Loss of GM-CSF signalling in non-haematopoietic cells increases NSAID ileal injury; furthermore, GM-CSF signalling in non-haematopoietic cells regulates ileal epithelial homeostasis via the STAT5 pathway. The therapeutic use of GM-CSF may therefore be beneficial in chronic ileitis associated with CD.
- Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF)
- Signals Transducers and Activators of Transcription (STAT5)
- Inflammatory Bowel Disease (IBD)
- NSAID
- Nonsteroidal Anti-inflammatory Drugs
- IBD basic research
- intestinal barrier function
- signal transduction
Statistics from Altmetric.com
- Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF)
- Signals Transducers and Activators of Transcription (STAT5)
- Inflammatory Bowel Disease (IBD)
- NSAID
- Nonsteroidal Anti-inflammatory Drugs
- IBD basic research
- intestinal barrier function
- signal transduction
Significance of this study
What is already known about this subject?
Administration of non-steroidal anti-inflammatory drugs (NSAIDs) is associated with frequent and early clinical relapse of quiescent inflammatory bowel disease and induced ileal injury.
Intestinal homeostasis is a critical component in the pathogenesis of diseases including food allergy and the inflammatory bowel diseases, which are rapidly increasing in frequency worldwide.
Administration of granulocyte-macrophage colony stimulating factor (GM-CSF) relieves symptoms in Crohn's disease (CD).
We have reported that GM-CSF bioactivity is associated with more aggressive ileal behaviour. GM-CSF-null mice exhibit ileal barrier dysfunction and develop a transmural ileitis following NSAID exposure.
What are the new findings?
Loss of GM-CSF signalling in non-haematopoietic cells increases NSAID ileal injury.
GM-CSF signalling in non-haematopoietic cells is essential for homeostatic intestinal barrier function.
GM-CSF signalling in non-haematopoietic cells regulates ileal epithelial homeostasis via activation of STAT5 in intestinal epithelial cells.
Our studies confer a novel mechanism for GM-CSF treatment of inflammatory bowel diseases by directly regulating apoptosis and proliferation of intestinal epithelial cells.
How might it impact on clinical practice in the foreseeable future?
Our studies have identified a GM-CSF-STAT5 pathway which directly regulates enterocyte function and response to injury relevant to human CD.
These studies will guide efforts to determine whether stimulation of intestinal barrier function via GM-CSF administration will be beneficial in restoring mucosal homeostasis in the subset of patients with CD with reduced GM-CSF bioactivity due to endogenous autoantibodies.
Our studies also suggested that the dose and duration of GM-CSF treatment should be cautiously considered due to a possible tumorigenic risk.
Introduction
Recent genome-wide association studies have suggested that defects in innate immunity contribute to the pathogenesis of Crohn's disease (CD).1 2 Granulocyte-macrophage colony stimulating factor (GM-CSF) promotes epithelial barrier integrity, stimulates myeloid cell antimicrobial function and reduces disease activity in CD and murine colitis due to dextran sodium sulfate administration.3–6 We have shown that patients with ileal CD have high levels of endogenous neutralising GM-CSF antibody in association with reduced GM-CSF bioactivity and neutrophil antimicrobial function.7 Administration of neutralising GM-CSF antibody to mice reduced ileal epithelial barrier function and increased susceptibility to NSAID-induced transmural ileitis in card15-deficient mice.7 The mechanisms by which GM-CSF may promote mucosal barrier function and adaptive responses to acute injury could involve direct effects on both non-haematopoietic and bone marrow (BM)-derived cells. GM-CSF has been shown to regulate the integrity of the alveolar epithelial barrier via direct effects on epithelial cells.8 Autocrine GM-CSF produced by freshly isolated murine intestinal epithelial cells (IEC) stimulated crypt cell proliferative activity.9 Systemic treatment with GM-CSF during hyperoxic exposure reduced epithelial apoptosis in the lungs.10 We therefore determined whether GM-CSF signalling in non-haematopoietic cells is required for intestinal homeostasis under basal conditions or in response to injury.
We have reported that STAT5 activation is reduced in the affected colon in CD and that stat5b-deficient mice exhibit increased susceptibility to experimental colitis due to trinitrobenzene sulfuric acid administration.11 12 We have demonstrated that STAT5b is required for basal survival of IEC by regulating the balance between pro- and anti-apoptotic gene expression.13 These studies have shown that STAT5 is required for intestinal homeostasis. It is also well known that STAT5 signalling is central to GM-CSF action in various cell types.14 15 Thus, the reduction in STAT5 activation which we have observed may reflect a reduction in local GM-CSF bioactivity.16 In our current studies, employing GM-CSF and GM-CSF ligand-deficient mice engrafted with wild type (WT) BM and gm-csf, gm-csfr and stat5 in vitro RNA interference, we demonstrate that GM-CSF signalling in non-haematopoietic cells is required for the maintenance of ileal homeostasis, and epithelial STAT5 is essential for GM-CSF regulation of homeostatic intestinal barrier function.
Methods
Materials
Human and mouse recombinant GM-CSF were obtained from R&D Systems (St Louis, Missouri, USA). All chemicals were purchased from Sigma-Aldrich (St Louis, Missouri, USA) unless otherwise noted. Antibodies specific for Bid, Bax, Bcl-2, GM-CSF, STAT5, p21 and cyclin D1 were from Santa Cruz Biotechnology (Santa Cruz, California, USA). Antibodies specific for cleaved caspase-3 (Asp175) and tyrosine phosphorylation specific STAT5 antibody (serine-649, pSTAT5) were from Cell Signaling Technology (Danvers, Massachusetts, USA). Antibodies specific for STAT5a and STAT5b were from Zymed Laboratories (South San Francisco, California, USA). Antibodies specific for CD3, Annexin V were from eBioscience (San Diego, California, USA). Another tyrosine phosphorylation specific STAT5 antibody was from Upstate Biotechnology (Lake Placid, New York, USA). ON-TARGET plus SMARTpool for human stat5a/b and gm-csfr α/βc were purchased from DHARMACON (Lafayette, Colorado, USA).13 The patient-based studies were approved by the CCHMC IRB and informed consent and assent were obtained from the study participants.7
Animal resources and maintenance
The animal study protocol was approved by the CHRF Institutional Animal Care and Use Committee (IACUC). Breeding pairs of gm-csf- and gm-csfrβc-deficient mice (GMKO and GMRKO) were obtained from Dr Bruce Trapnell.7 All mice used in these studies were re-genotyped. Mice were maintained in specific pathogen-free (SPF) conditions in the Children's Hospital Research Foundation (CHRF) Animal Care Facility.
Murine bone marrow transplantation (BMT)
This technique was performed with the assistance of the CHRF Experimental Haematology Mouse Bone Marrow Transplantation (BMT) Core. The BMT core ablated the BM with whole-body gamma irradiation with 1200 Rad in divided doses 3 h apart. The recipient then immediately received an intravenous tail vein injection of 1×106 whole BM donor cells. As shown in table 1, we transplanted WT BM cells into WT GMKO and GMRKO recipients (WTBM, WTBM→GMKO and WTBM→GMRKO mice, respectively) or transplanted GMRKO BM cells into WT mice (GMRBM→WT mice). Controls included gender and age-matched WT, GMKO and GMRKO mice without BMT. We tested the transplantation efficiency by measuring CD45.1 and CD45.2 in blood and BM; in parallel, we used flow cytometry (FACS) and immunofluorescence (IF) to detect gm-csf and gm-csfrβc expression in peripheral blood, BM and ileal sections, respectively (see figure 2A–C in online supplement). Reconstitution of >95% was anticipated. Eight weeks after BMT we determined the effect on ileal barrier function and susceptibility to NSAID-induced ileitis.
Murine bone marrow transplantation (BMT)
Animal models of ileitis
To establish the animal model of ileitis, 4- or 5-week-old male and female mice were exposed to the NSAID piroxicam at 200 ppm in the chow for 2 weeks. Ileitis histology was scored using the method previously published.7 17
Primary mouse IEC short-term culture
Briefly, mouse ileum was everted and incubated in calcium-magnesium-free HBSS with 1 mM EDTA for 30 min at 37°C with gentle shaking to liberate IEC. Cell survival was determined with Annexin V kit (eBioscience). The isolated cells were seeded in 24-well Matrigel coated plates (BD Bioscience) and subconfluent IEC were exposed to 1 ng/ml recombinant mouse GM-CSF for 30 min.7 18 The details are described in the online supplement.
Measurement of paracellular ileal permeability and bacterial translocation
Ileal and colonic paracellular permeability were determined using an everted gut sac and Ussing chambers method.7 19 20 The details are described in the online supplement.
Immunoblotting and immunofluorescence
Expression of GM-CSF, GM-CSFRα and βc, cleaved caspase-3, bax, Bid, p21, cyclin D1 and STAT5 were measured with isolated ileal IEC, ileal mucosal tissue and cultured cells.7 21 The details are described in the online supplement.
BrdU labelling and ki-67, TUNEL and cleaved caspase 3 staining of tissue sections and semiquantitative analysis of IEC protein expression
BrdU staining followed the manufacturer's instructions (BrdU In-Situ Detection Kit, BD Pharmingen).21 The details are given in the online supplement.
FACS analysis of IEC apoptosis
CD3-/7-AAD-/AnnexinV+ was used as a marker for apoptotic cells (BD Biosciences).7 22 Data were analysed using FlowJo software. The details are given in the online supplement.
RNA interference
gm-csf, gm-csfr α and βc and stat5 a and b ON-TARGETplus SMARTpool (Dharmacon, Chicago, Illinois, USA) transfection were performed with preconfluent HT-29 and Caco-2 cells according to the manufacturer's instructions. Subconfluent cells were then co-incubated with 10−4 M indomethacin (Sigma) and 1 ng/ml recombinant human GM-CSF (rhGM-CSF, R&D Systems) for 24 h or treated with 10 ng/ml recombinant human tumour necrosis factor α (rhTNFα, R&D Systems) in the presence or absence of stat5 a and b or gm-csfrαc and βc siRNA.13 The details are described in the online supplement.
Measurement of IEC proliferation and GM-CSF level in serum, isolated cells and tissue extracts
The details are described in the online supplement.
Statistical analysis
The results are presented as mean±SEM. Data were analysed using analysis of variance followed by one-way ANOVA and a two-tailed Student t test; p values <0.05 were considered significant.
Results
Increased levels of GM-CSF autoantibodies (GM-CSF) in ileal CD are associated with dysregulation of IEC survival and proliferation
We found that the frequency of apoptotic ileal crypt epithelial cells was increased in biopsy sections obtained from inflamed ileal mucosa in paediatric patients with ileal CD with elevated GM-CSF autoantibodies (defined as ≥1.6 μg/ml,7 figure 1A) compared with patients with CD with low GM-CSF autoantibodies and healthy controls. It should be noted that the severity of ileal mucosal inflammation was also substantially higher in patients with elevated GM-CSF autoantibodies (4.5±0.9) compared with 2.2±1.0 in the group with low GM-CSF autoantibodies and 0 in the healthy control group.7 23 Despite the overall increase in mucosal injury, the frequency of proliferating ileal crypt epithelial cells was reduced in the inflamed area in patients with CD with elevated GM-CSF autoantibody levels compared with those with low GM-CSF autoantibody levels and healthy controls (figure 1B). Collectively, these data suggest that GM-CSF autoantibodies may promote more aggressive ileal disease in part by suppression of intestinal epithelial function in response to gut injury.

Increased levels of granulocyte-macrophage colony stimulating factor (GM-CSF) autoantibody (Ab) in Crohn's disease (CD) are associated with dysregulation of survival and proliferation of intestinal epithelial cells. (A) The frequency of apoptotic ileal crypt epithelial cells was determined by cleaved caspase-3 staining in healthy controls (CON) and patients with CD with high (Hi) or low (Lo) serum GM-CSF Ab. Results are expressed as mean±SEM (n=7); original magnification ×200; bar=50 μm. (B) Frequency of proliferative ileal crypt epithelial cells was determined by Ki67 staining in healthy controls and patients with CD with high or low serum GM-CSF Ab. Results are expressed as mean±SEM (n=7); original magnification ×400; bar=50 μm.
Loss of GM-CSF signalling in non-haematopoietic cells increases susceptibility to NSAID-induced ileitis
The contribution of cells from haematopoietic versus non-haematopoietic lineages in the pathogenesis of CD is unclear. Studies have shown that haematopoietic cell lineages such as neutrophils, macrophages and lymphocytes play an important role in the pathogenesis of CD.24 Conversely, recent studies have also shown that non-haematopoietic sources, especially IEC barrier dysfunction, are important aetiological factors contributing to CD.25 Thus, dysregulated IEC barrier function may be a prerequisite for the onset of intestinal inflammation.25 26 We first determined that an appreciable amount of GM-CSF was expressed in mouse ileal IEC and the intestinal mucosa under basal conditions (see figure 1A in online supplement). This was confirmed by ELISA assay, as the GM-CSF concentration was 62±1.0 pg/ml in the serum compared with 2.6±0.5 pg/ml and 5.06±0.8 pg/ml in the same amount of isolated ileal IEC and ileal mucosa, respectively. IF and FACS analysis (see figures 1B and 2A in online supplement) with isolated IEC confirmed that GM-CSFRβc is highly expressed on ileal IEC compared with endothelial cells and colonic IEC.
We next evaluated whether GM-CSF signalling in non-haematopoietic cells is mainly involved in this regulation. Murine gm-csfrβc deficiency disrupts both GM-CSF and interleukin 5 (IL-5) signalling in BM-derived cells but will allow for IL-3 function via the IL-3β subunit.27 Importantly, IEC do not express the cognate IL-5 receptor, so gm-csfrβc deficiency results in a specific depletion in GM-CSF signalling, relative to WT controls, in IEC.28 29 We therefore generated BM chimeras by reconstituting irradiated GM-CSF receptor (gm-csfr) β chain or GM-CSF-deficient mice with WT BM (WTBM→GMRKO and WTBM→GMKO) and WT controls containing WT BM transplanted from WT donors (WTBM). Although a relatively small amount of other non-haematopoietic cells beyond IEC also lost gm-csfrβc expression and BM-derived cells may transdifferentiate into epithelial cells in response to injury,30 31 WTBM→GMRKO and WTBM→GMKO chimeras still showed a considerable loss of GM-CSF or GM-CSF signalling in IEC which account for the majority of intestinal mucosal cells. IF and FACS analysis confirmed reconstitution of GM-CSF+ and GMCSFRβc+ lamina propria mononuclear cells (LPMC) and circulating leucocytes, respectively, in WTBM→GMRKO chimeras and WTBM→GMKO chimeras. Most importantly, IF confirmed the depletion of gm-csfrβc on IEC (see figure 2A–C in online supplement). WTBM→GMRKO may therefore represent a model for studying GM-CSF signalling in non-haematopoietic (eg, epithelial) cells.
We then determined whether loss of GM-CSF signalling or GM-CSF in non-haematopoietic cells would increase susceptibility to NSAID-induced ileitis. Similar to piroxicam-treated gm-csf-deficient mice (GMKO)7 and gm-csf receptor β chain-deficient mice (GMRKO), we found that a significant ileitis developed in WTBM→GMRKO chimeras following a 2-week exposure to piroxicam (figure 2A). In contrast, BM chimeras containing deletion of gm-csfrβc within the BM-derived compartment (GMRBM→WT) did not develop significant ileal disease following administration of piroxicam (figure 2A–C). Together these data indicate that the defective GM-CSF signalling in the non-haematopoietic compartment but not in the transplanted haematopoietic cells was sufficient for NSAID-induced ileitis in mice.

Loss of granulocyte-macrophage colony stimulating factor (GM-CSF) signalling in non-haematopoietic cells increases susceptibility to non-steroidal anti-inflammatory drug (NSAID)-induced ileitis. (A–C) WT, GMKO, GMRKO, WTBM, WTBM→GMKO, GMRBM→WT and WTBM→GMRKO mice received piroxicam (PIR) in the chow for 2 weeks and the effects on ileal histopathology and ulceration were determined. Original magnification ×100, bar=50 μm. Open circles represent the ulcer area. *p<0.01 vs WTBM; #p<0.01 vs GMKO (n=10). (D) Bacterial translocation in mesenteric lymph nodes (MLN) was determined in WT, WTBM, GMKO, GMRKO, WTBM→GMKO, GMRBM→WT and WTBM→GMRKO mice with and without piroxicam administration. *p<0.05 vs non-PIR WT mice; #p<0.05 vs non-PIR WTBM and WTBM→GMKO mice (n=10). Results are expressed as mean±SEM.
Restoration of WTBM improves NSAID-induced ileitis in GM-CSF deficient mice
While we found that WTBM→GMKO chimeras also developed ileitis in the intestinal mucosa following exposure to piroxicam, the disease severity as well as ileal ulceration was significantly milder than in the corresponding GMKO and WTBM→GMRKO chimeras (figure 2A–C). It indicated that BM-derived cells also had a role in the protection against ileal injury; restoration of WTBM improved NSAID-induced ileal injury in GMKO but not in GMRKO mice. Consistent with these differences in mucosal injury, we found that there was significantly increased bacterial translocation into mesenteric lymph nodes in WTBM→GMRKO, WTBM→GMKO, GMKO and GMRKO mice following piroxicam exposure compared with WTBM and WT controls (figure 2D). Taken together, these data indicate that GM-CSF signalling in non-haematopoietic cells is required for homeostatic responses to mucosal injury.
Loss of GM-CSF signalling in non-haematopoietic cells leads to ileal barrier dysfunction
To investigate whether loss of GM-CSF signalling in non-haematopoietic cells may be responsible for the ileitis susceptibility trait in GMRKO and WTBM→GMRKO mice, we determined ileal and colonic paracellular permeability. Ileal but not colonic paracellular permeability was significantly increased in GMKO, GMRKO and WTBM→GMRKO mice (see figure 3A and figure 3A–C in online supplement), which led to significant bacterial translocation to MLN under basal conditions (figure 2D). Consistent with this, transepithelial electric resistance (TEER) was significantly lower in the ileum but not in the colon of WTBM→GMRKO chimeras (see figure 3B and figure 3D in online supplement). Interestingly, ileal paracellular permeability in WTBM→GMKO chimeras was significantly reduced compared with ileal permeability in GMKO mice (p<0.01, figure 3A), suggesting that WT BM-derived cells may rescue the ileal barrier dysfunction due to loss of GM-CSF in intestinal mucosa and further improve the disease severity of NSAID-induced ileitis (figure 2A–C). Most importantly, it confirmed that GM-CSF signalling in non-haematopoietic cells is required for ileal epithelial barrier function.
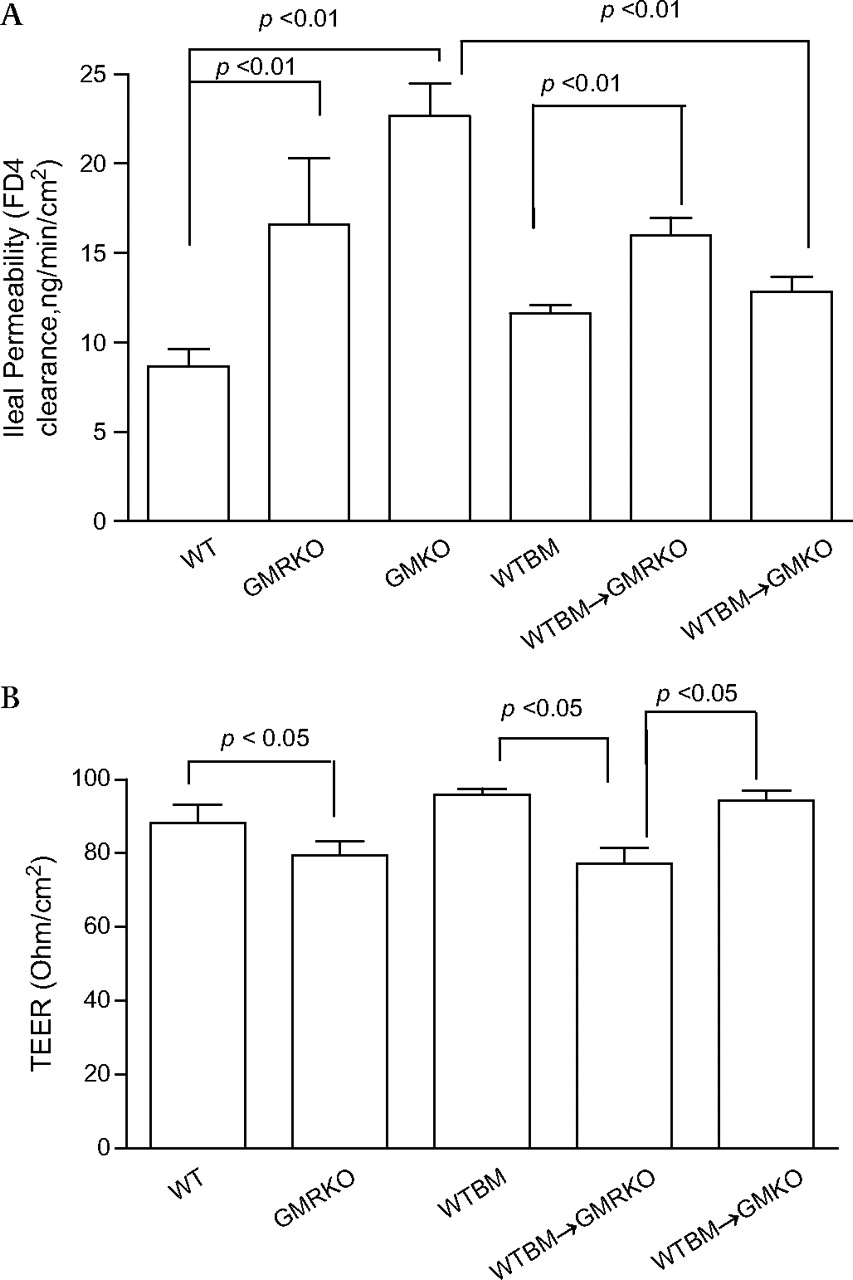
Loss of granulocyte-macrophage colony stimulating factor signalling in non-haematopoietic cells leads to intestinal barrier dysfunction. (A) Ileal paracellular permeability was determined with everted gut sac in wild type (WT), WTBM, GMKO, WTBM→GMRKO and WTBM→GMKO mice (n=10). (B) Ileal epithelial electric resistance (TEER) was determined by clamping the voltage across the epithelium layer mounted in Ussing chambers (n=5). Results are expressed as mean±SEM.
GM-CSF signalling in non-haematopoietic cells is required for intestinal epithelial survival response to injury
The frequency of apoptotic IEC, as measured by TUNEL and activated caspase-3 immunohistochemistry staining, did not differ under basal conditions in GMKO and GMRKO mice relative to WT controls or in WTBM→GMKO and WTBM→GMRKO chimeras compared with WTBM. This therefore excluded the possibility of irradiation-induced epithelial injury. However, the frequency of apoptotic IEC was significantly increased in GMKO compared with WT mice following exposure to piroxicam (see table 2, figures 4A and B). Interestingly, we found that WTBM→GMKO chimeras had a significantly reduced frequency of IEC apoptosis following exposure to piroxicam compared with GMKO and WTBM→GMRKO chimeras (table 2, figure 4A and B), suggesting that blockade of GM-CSF signalling resulted in apoptosis of IEC.
Effects of GMKO, GMRKO and different bone marrow chimeras on apoptosis of ileal intestinal epithelial cells (IEC)



Granulocyte-macrophage colony stimulating factor (GM-CSF) is required for intestinal epithelial survival response to injury. (A) TUNEL and cleaved caspase-3 immunohistochemistry staining were performed on ileal sections from WT, GMKO, WTBM, WTBM→GMKO and WTBM→GMRKO mice under basal conditions and following exposure to piroxicam (PIR). Average apoptotic intestinal epithelial cells (IEC) per crypt was determined (original magnification ×400, bar=50 μm). Ileal epithelial cells were isolated with 1 mM EDTA from GMKO, WTBM, WTBM→GMKO and WTBM→GMRKO mice with and without piroxicam treatment. (B, C) IEC apoptosis was determined by cleaved caspase-3 western blot analysis (n=7). (D) The frequency of apoptotic IEC was determined as the 7-AAD−, CD3−, Annexin V+ population (n=7). (E) Bax and Bcl-2 expression were examined by western blot analysis (n=7). Signal intensity was determined by densitometry. Results shown as mean±SEM. NSAID, non-steroidal anti-inflammatory drug.
However, while ileal IEC survival did not vary under basal conditions, cleaved caspase-3 in isolated ileal IEC was upregulated in WTBM→GMRKO mice compared with WTBM mice following exposure to piroxicam (table 2, figures 4A and C). Concomitant with this, the frequency of apoptotic IEC (identified by Annexin V FACS and cleaved caspase-3 immunohistochemistry staining) was increased in piroxicam-treated WTBM→GMKO mice (table 2, figure 4D). Furthermore, we confirmed that the bax/bcl-2 ratio was significantly higher in isolated ileal IEC from piroxicam-treated WTBM→GMKO mice compared with piroxicam-treated WTBM mice (figure 3E). These results show that GM-CSF signalling in non-haematopoietic cells is required for intestinal epithelial survival response to injury.
GM-CSF signalling in non-haematopoietic cells regulates intestinal epithelial cellular proliferation
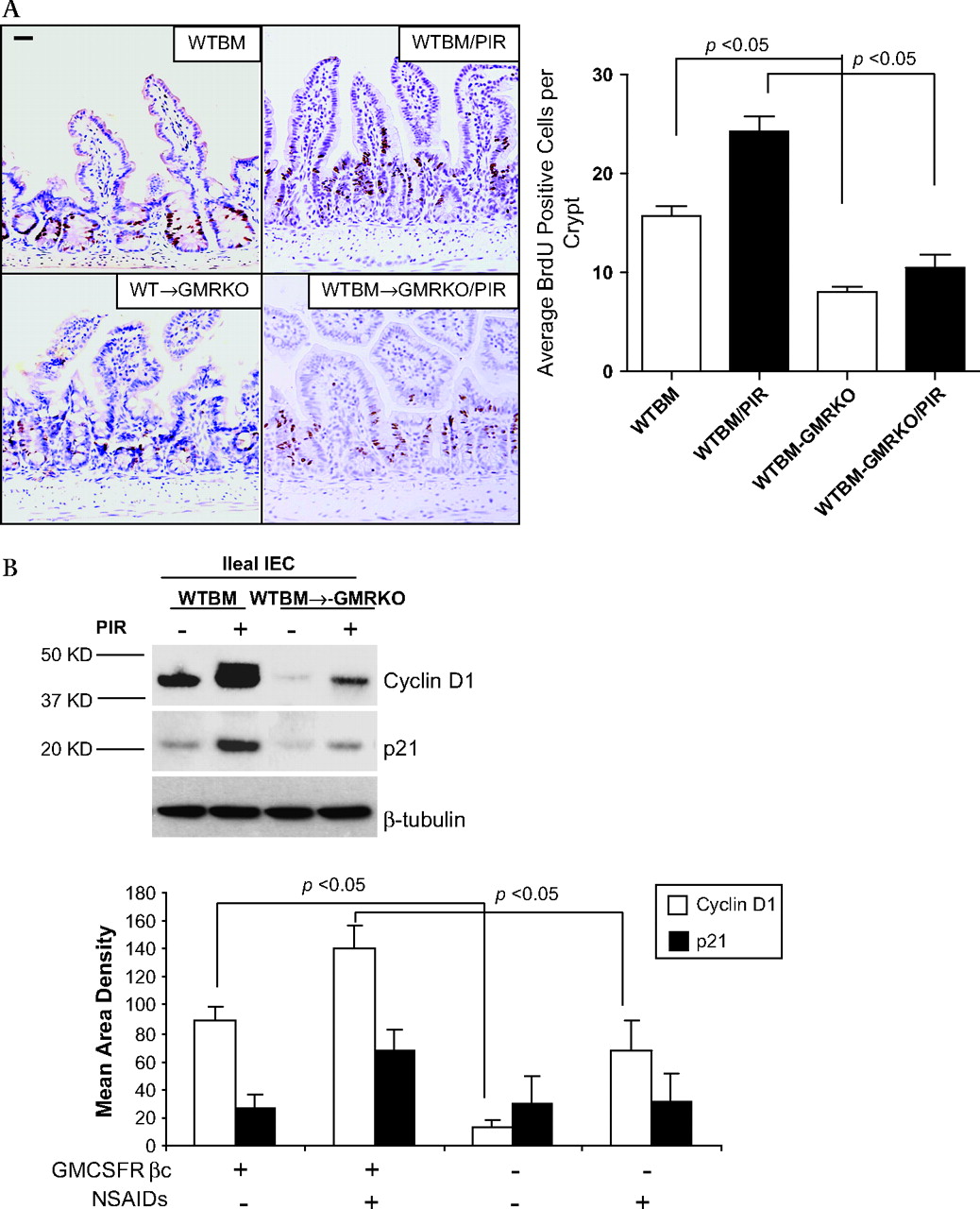
To address this further, we examined ileal IEC proliferation as measured by BrdU labelling. IEC proliferation was substantially reduced in WTBM→GMRKO chimeras, accompanied by significantly lower levels of p21 and cyclin D1 expression, compared with WTBM controls assessed under basal conditions (figures 5A and B). Interestingly, the BrdU labelling study showed that piroxicam administration increased the migration of ileal IEC with upregulation of cyclin D1 and p21 in WTBM mice; this effect was greatly abrogated in WTBM→GMRKO mice (see figures 5A and B). GM-CSF signalling on ileal IEC is therefore required for cellular proliferation under basal conditions and following NSAID exposure. In contrast, loss of GM-CSF signalling on ileal IEC was sufficient to cause a reduction in ileal IEC proliferation which may initiate the ileal barrier dysfunction. Collectively, these studies confirm that GM-CSF signalling in non-haematopoietic cells is required for ileal homeostasis under basal conditions and following NSAID exposure.

Granulocyte-macrophage colony stimulating factor (GM-CSF) signalling in non-haematopoietic cells regulates cellular proliferation. (A) Proliferation of ileal intestinal epithelial cells (IEC) was determined using BrdU incorporation measured by immunohistochemistry staining (n=7 mice, ≥200 crypts counted per group). Original magnification ×400, bar=50 μm. (B) Ileal IEC were isolated with 1 mM EDTA from WTBM and WTBM→GMRKO mice with and without piroxicam (PIR) treatment, cyclin D1 and p21 were determined by western blot analysis (n=5). Results shown as mean±SEM. NSAID, non-steroidal anti-inflammatory drug.
GM-CSF regulates IEC survival and proliferation in vitro
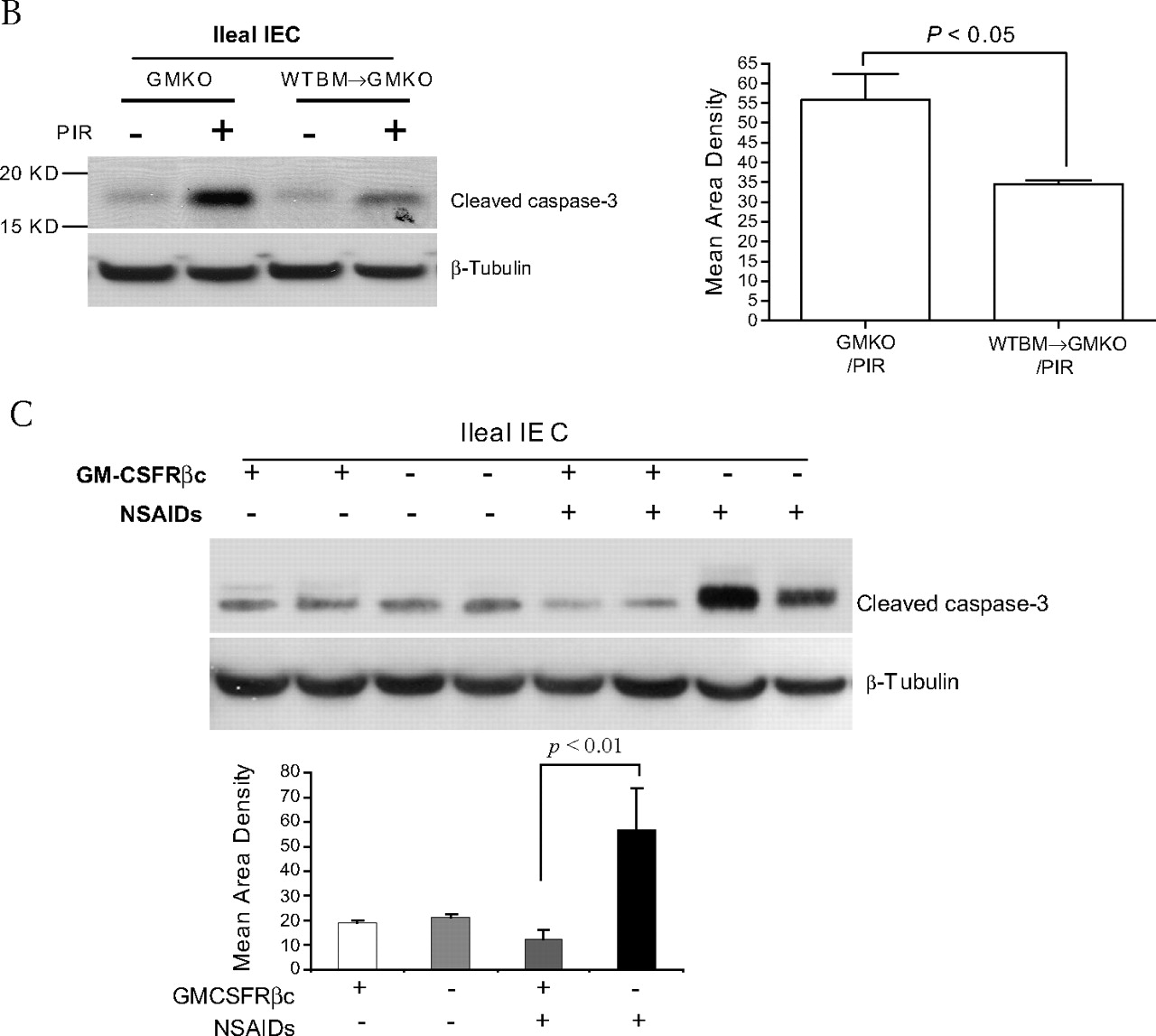
In vitro studies showed that GM-CSF at 1 ng/ml normalised proliferation of subconfluent Caco-2 and HT-29 cells exposed to indomethacin (figure 6A). We significantly knocked down the expression of GM-CSF in subconfluent Caco-2 cells and found that the proliferation of Caco-2, determined by BrdU incorporation, was significantly suppressed by the reduction of GM-CSF. The proliferation was restored by the reintroduction of GM-CSF (1 ng/ml) into the culture medium (figure 6B). Similar to our in vivo results, in vitro GM-CSF knock-down also induced significant activation of caspase-3 in Caco-2 cells (figure 6C).

Granulocyte-macrophage colony stimulating factor (GM-CSF) regulates survival and proliferation of intestinal epithelial cells in vitro. (A) Subconfluent Caco-2 and HT-29 cells were incubated with rhGM-CSF (1 ng/ml) and indomethacin (IN, 10−4 M) for 24 h, cell proliferation was measured using BrdU incorporation. *p<0.01 vs control (CON) group; #p<0.01 vs IN group (n=10). (B, C) gm-csf siRNA was used to knock down the expression of GM-CSF in subconfluent Caco-2 cells in the absence and presence of rhGM-CSF (1 ng/ml) administration for 24 h. Proliferation of Caco-2 cells was detected by BrdU incorporation and apoptosis of Caco-2 cells by cleaved caspase-3 western blot analysis. Control (CON) group: transfection reagent without siRNA; *p<0.05 vs control or GM-CSF group (n=10). (D) Proliferation of Caco-2 cells was detected under gm-csfαc or βc knock-down. *p<0.01 vs control group (n=10). Percentage of knock-down was determined by western blot analysis. (E) Cyclin D1 and p21 were determined by western blot analysis in subconfluent Caco-2 and HT-29 cells with and without gm-csfrβc knock-down. (F) After 24 h tumour necrosis factor α (TNFα) (10 ng/ml) administration, cleaved caspase-3 was measured by western blot analysis in subconfluent Caco-2 cells with and without gm-csfrβc knock-down. Results representative of five independent experiments are shown. BrdU incorporation results are shown as mean±SEM.
Both gm-csfr α and βc knock-down reduced basal cell proliferation in subconfluent Caco-2 and HT-29 cells, and these cells were more susceptible to the suppressive effects of indomethacin or TNFα on proliferation (figure 6D and data not shown). The abundance of cyclin D1 and p21 was reduced following gm-csfrβc knock-down, which is consistent with the variation in cellular proliferation (figure 6E). As with our in vivo studies, TNFα upregulated cleaved caspase-3 in subconfluent Caco-2 cells, which was enhanced by gm-csfrβc knock-down (figure 5F). Knock-down of either GM-CSF or its receptor in intestinal cell lines resulted in suppression of proliferation and an increase in apoptosis, showing that GM-CSF can directly regulate IEC proliferation and survival via an autocrine pathway.
GM-CSF activates STAT5 in IEC in response to injury
GM-CSF executes its functions via activation of the JAK2-STAT5 signalling pathway.32 We found that STAT5 activation was increased in IEC and LPMC of WTBM mouse ileum following piroxicam administration. This response was abrogated in the IEC but not LPMC of WTBM→GMRKO mice. Interestingly, epithelial STAT5 activation was well preserved in WTBM→GMKO mice, suggesting the effect of GM-CSF from BM-derived cells on IEC (figure 7A and B). In fact, the activation of STAT5 in LPMC appeared to be increased further in WTBM→GMRKO mice after piroxicam administration (figure 7A). GM-CSFRβc is known to mediate haematopoietic cell survival through induction of the anti-apoptotic protein bcl-2.33 We therefore examined whether GM-CSF could directly regulate enterocyte survival and proliferation in response to injury via activation of STAT5. We isolated ileal IEC and 1 ng/ml GM-CSF was used to stimulate the cells for 30 min. We found that GM-CSF activated STAT5 in WT ileal IEC. Most importantly, GM-CSF upregulated bcl-2 expression in WT ileal IEC but not in GM-CSFRβc-deficient ileal IEC (figure 7C and D and data not shown). These data suggest that GM-CSF may regulate ileal IEC survival via a GM-CSF:STAT5:Bcl-2 pathway.

Granulocyte-macrophage colony stimulating factor (GM-CSF) activates STAT5 in intestinal epithelial cells (IEC) in response to injury. (A) WTBM and WTBM→GMRKO mice received piroxicam (PIR) in the chow for 2 weeks and STAT5 activation was examined by immunohistochemistry staining (original magnification ×100, bar=50 μm, inset is negative control). Ileal epithelial cells in WTBM, WTBM→GMKO and WTBM→GMRKO mice with and without proxicam treatment were isolated. (B) pSTAT5 and STAT5 in nuclear proteins (NE) was determined by western blot analysis. (C, D) Ileal IEC were isolated from WT mice and then stimulated with rmGM-CSF (1 ng/ml) for 30 min; pSTAT5 in NE and Bcl-2 in cytosolic protein (CE) were detected by western blot analysis. Signal intensity was determined by densitometry and results are shown as mean±SEM (n=5).
GM-CSF regulates the survival and proliferation of IEC cells through activation of STAT5 in vitro
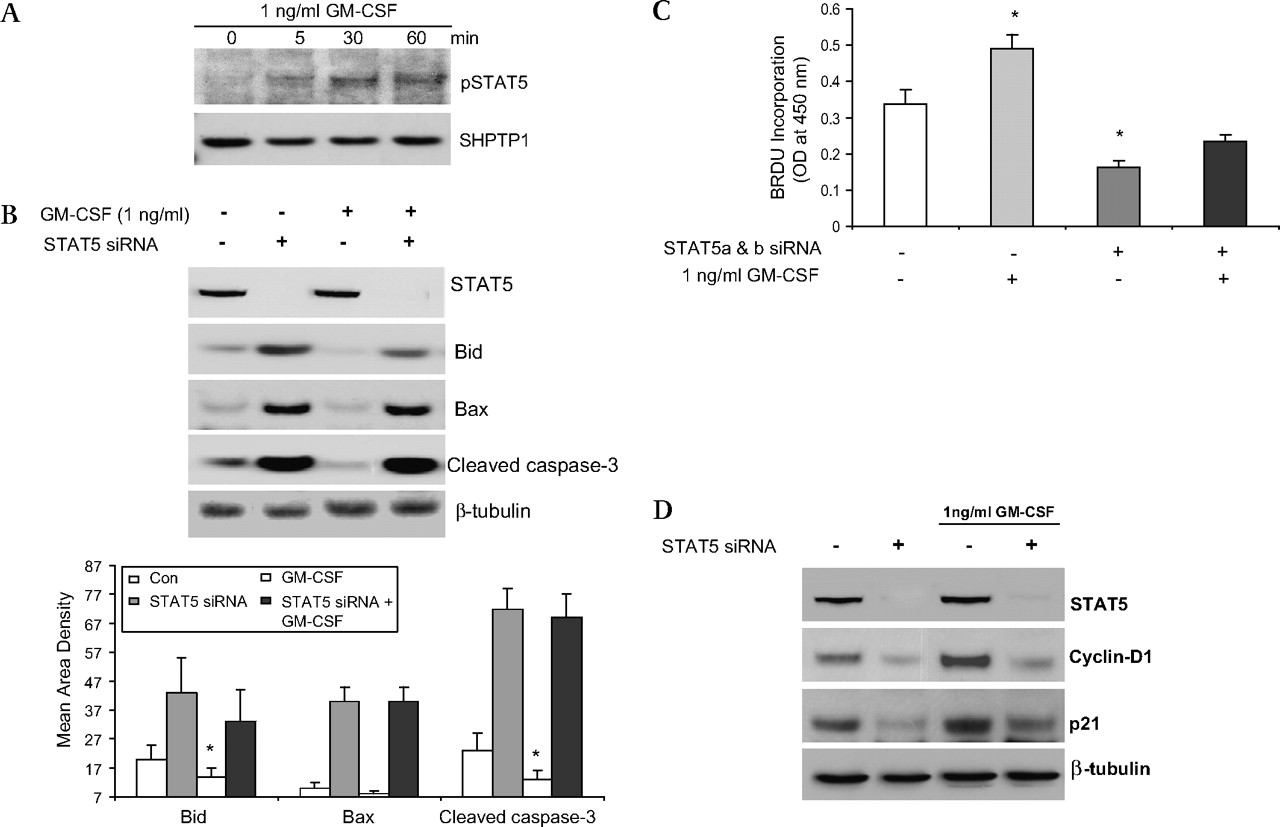
We exposed subconfluent Caco-2 cells to GM-CSF (1 ng/ml) and found that GM-CSF can induce tyrosine phosphorylation of STAT5 (figure 8A). We next used RNAi to knock down the expression of both stat5a and stat5b (STAT5 RNAi) in Caco-2 cells and measured markers of apoptosis and proliferation. We also found that stat5 knock down greatly increased the amount of Bid, Bax and cleaved caspse-3. Treatment with GM-CSF (1 ng/ml) reduced the level of cleaved caspase-3 as well as Bid under basal conditions; however, this effect was impeded by addition of STAT5 RNAi (figure 8B). We then examined proliferation by measuring BrdU incorporation and levels of cyclin D1 and p21. Treatment with GM-CSF increased Caco-2 proliferation and cyclin D1 and p21 expression; these effects were abrogated by STAT5 knock down (figures 8C and D). These data clearly indicate that GM-CSF regulates the survival and proliferation of IEC cells through activation of STAT5.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Granulocyte-macrophage colony stimulating factor (GM-CSF) regulates the survival and proliferation of intestinal epithelial cells through activation of STAT5. (A) Subconfluent Caco-2 cells were stimulated with rhGM-CSF (1 ng/ml) for 15, 30 and 60 min and the activation of STAT5 (pSTAT5) was examined in nuclear proteins (NE) by western blot analysis. (B) STAT5, Bid, Bax and cleaved caspse-3 expression were measured by western blot analysis with and without STAT5 a and b siRNA (STAT5 siRNA). Control (CON) group: transfection reagent without siRNA. (C) Caco-2 cells were stimulated by rhGM-CSF (1 ng/ml) with and without STAT5 siRNA and cell proliferation was measured using BrdU incorporation. *p<0.05 vs control group. (D) Cyclin D1 and p21 were identified by western blot analysis with and without STAT5a and b RNAi. Signal intensity was determined by densitometry and is shown as the mean±SEM. Results representative of five independent experiments are shown.
Discussion
Mechanisms by which GM-CSF may promote mucosal barrier function in the intestine have only recently begun to be explored.6 7 It may involve both non-haematopoietic and BM-derived cells. gm-csfrβc is highly expressed on ileal IEC and specifically conducts the GM-CSF signalling in IEC.27 This indicates that GM-CSF signalling in non-haematopoietic cells such as epithelial cells might be required for ileal function. We have now shown that GM-CSF signalling in non-haematopoietic cells regulates the homeostasis of intestinal barrier integrity. A key mechanism in the therapeutic effect of GM-CSF on patients with CD may therefore involve GM-CSF regulation of IEC function.
NSAID administration is associated with frequent and early clinical relapse of quiescent inflammatory bowel disease and induced ileal injury.34 Our data indicated that GM-CSF signalling as well as GM-CSF deficiency lead to a disrupted ileal barrier and increased susceptibility to NSAID-induced ileitis. To study the importance of GM-CSF signalling on ileal barrier function we therefore used WTBM→GMRKO chimeras which have a specific reduction of GM-CSF signalling in non-haematopoietic cells relative to WT controls. We found that significant ileitis developed in these WTBM→GMRKO mice but not in GMRBM→WT mice following exposure to piroxicam for 2 weeks. In comparison, transplantation of BM from WT donor mice to gm-csf-deficient recipient mice prevented NSAID-induced ileal injury and restored ileal barrier function and STAT5 activation in IEC. On the one hand these results confirmed the importance of GM-CSF signalling in non-haematopoietic cells in promoting the enterocyte response to NSAID injury while, on the other hand, it also indicated that the BM-derived compartment may be the main source of GM-CSF in the intestine. GM-CSF might have an anti-inflammatory role in the maintenance of homeostatic ileal barrier function through directly targeting IEC.
Cyclin D1 and p21 regulate the intestinal cell cycle and proliferative activity in response to injury.35 We found that the expression of cyclin D1 and p21 was lower in gm-csfrβc-deficient IEC than in WT IEC and was less responsive to NSAID injury in WTBM-GMRKO chimeras. It is therefore plausible to conclude that a lack of sufficient cyclin D1 and p21 upregulation leads to reduced proliferative activity in response to injury which then results in a greater susceptibility to NSAID-induced injury. Consistent with in vivo studies, we found that subconfluent Caco-2 and HT-29 cells with reduced gm-csfrαc or gm-csfrβc expression due to RNAi exhibited reduced proliferation under basal conditions and enhanced apoptosis induced by TNFα. These studies indicate that GM-CSF is required for IEC proliferation and survival. GM-CSF signalling is therefore more likely to have an anti-inflammatory role in enterocytes in response to piroxicam administration. Our findings reveal a novel mechanism for GM-CSF treatment of inflammatory bowel disease by directly regulating IEC apoptosis and proliferation. Conversely, our studies also suggest that the dose and duration of GM-CSF treatment should be cautiously considered due to a possible tumorigenic risk.36 Taken together, these results confirm that GM-CSF signalling in non-haematopoietic cells is required for ileal homeostasis, both under basal conditions and following NSAID exposure.
GM-CSF executes its functions by interacting with the corresponding receptor which phosphorylates at least two distinct domains in the β-chain.37 One of these domains induces the activation of mitogen-activated protein kinases and the PI-3K/Akt/p21waf-1 pathway. The other mediates the activation of the JAK2-STAT5 signalling pathway.32 STAT5 activation is the central pathway for GM-CSF regulation of proliferation and survival of various cells.16 Activation of STAT5 is required for the proliferation, differentiation and maintenance of secretory mammary epithelium, hepatocytes and regulatory T cells. STAT5 upregulates the transcription of antiapoptotic bcl family members in mammary epithelial cells, haematopoietic cells and cardiac myocytes.38 39 GM-CSF βc is directly associated with JAK2 which sequentially activates phosphorylation of downstream STAT5 in response to different concentrations of GM-CSF.15 40 STAT5a and b can also bind the distal GAS site in the cyclin D1 promoter to regulate the cell cycle.41 We found that exogenous GM-CSF activated STAT5 as well as STAT5-regulated downstream genes including bcl-2 in murine primary IEC. STAT5 RNAi led to the suppression of Caco-2 cell proliferation and blocked the ability of GM-CSF to stimulate proliferation. In vivo, chronic piroxicam administration upregulated STAT5 activity in IEC in WTBM and WTBM→GMKO mice; this was abrogated in WTBM→GMRKO mice in which GM-CSF signalling in non-haematopoietic cells was significantly blocked. Collectively, these data strongly suggest that GM-CSF regulation of IEC proliferation requires the activation of STAT5. Our current studies employed a Cre/loxP recombination system to specifically delete the Stat5 locus in IEC in mice.42 Depletion of STAT5 in IEC increased apoptotic IEC and reduced IEC proliferation at baseline (data not shown). Taken together, these studies confirm that enterocyte STAT5 is essential for ileal epithelial cell homeostasis and indicate that GM-CSF regulates the survival and proliferation of IEC cells through activation of STAT5.
In conclusion, our studies have identified a GM-CSF-STAT5 pathway which directly regulates enterocyte function and response to injury relevant to human CD. These studies will guide efforts to determine whether stimulation of intestinal barrier function via GM-CSF administration will be beneficial in restoring mucosal homeostasis in the subset of patients with CD with reduced GM-CSF bioactivity due to endogenous autoantibodies.
Acknowledgments
The authors acknowledge the assistance of the CHRF Experimental Haematology Mouse BMT Core and thank Drs Lee Denson, Elizabeth Mann and Mitchell Cohen for revising the manuscript and providing outstanding technical assistance.
References
Supplementary materials
Web only data
Files in this Data Supplement:
Footnotes
Funding Crohns and Colitis Foundation of America (XH), Cincinnati Children's Hospital Research Foundation Digestive Health Center (PHS Grant P30 DK0789392) and Cincinnati Children's Hospital Medical Center Trustee Award (XH). NIH clinical and translational research award KL2 RR026315 (XH).
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Cincinnati Children's Hospital Medical Center, IRB #04-1-10.
Provenance and peer review Not commissioned; externally peer reviewed.