





Abstract
To understand more fully how amino acid composition of proteins has changed over the course of evolution, a method has been developed for estimating the composition of proteins in an ancestral genome. Estimates are based upon the composition of conserved residues in descendant sequences and empirical knowledge of the relative probability of conservation of various amino acids. Simulations are used to model and correct for errors in the estimates. The method was used to infer the amino acid composition of a large protein set in the Last Universal Ancestor (LUA) of all extant species. Relative to the modern protein set, LUA proteins were found to be generally richer in those amino acids that are believed to have been most abundant in the prebiotic environment and poorer in those amino acids that are believed to have been unavailable or scarce. It is proposed that the inferred amino acid composition of proteins in the LUA probably reflects historical events in the establishment of the genetic code.
Introduction
An insight into how proteomic amino acid composition has changed over vast evolutionary time is required for a thorough understanding of the process of protein evolution. Knowledge of the amino acid composition of early proteomes can reveal which of the amino acids have increased and which have decreased in frequency with evolution. Such information could provide clues to the order in which amino acids were introduced into the genetic code and thus into primitive proteins, as suggested previously on the basis of a different approach (Brooks and Fresco 2002 ).
The recent increase in the number of whole genome sequences has made the analysis of the corresponding inferred proteomes possible. These analyses have included a statistical description of amino acid composition and sequence length (Gerstein 1998a ), and surveys of both structurally determined (Gerstein 1998b;Wolf, Grishin, and Koonin 2000 ) and predicted folds (Gerstein 1997 ). Interspecies proteomic comparisons have revealed the phylogenetic distribution of protein families (Tatusov, Koonin, and Lipman 1997 ); in turn, this knowledge has been used to infer the protein complement of the Last Universal Ancestor (LUA) (Kyrpides, Overbeek, and Ouzounis 1999 ), the single-celled ancestor that gave rise to the three primary lineages, the eubacteria, archaea, and eukaryotes.
Here, we extend these earlier proteomic characterizations by estimating the amino acid composition of proteins in an ancestral genome. The existing methods used for the reconstruction of ancestral protein sequences, maximum parsimony (MP) and maximum likelihood (ML), are by themselves insufficient to provide accurate estimates of ancestral sequence composition. Where sequences have diverged significantly, MP can only provide partial sequence reconstructions; the identity of residues at many sites in the ancestral sequence will remain ambiguous. Moreover, those sites with assigned residues will be biased toward amino acids that tend to be conserved. On the other hand, ML requires an assumption of the amino acid composition of the sequences being reconstructed, and it is usually assumed that the composition of the ancestors is the same as that of the extant descendants (Yang, Kumar, and Nei 1995 ). Although Galtier, Tourasse, and Gouy (1999) estimated the GC content of ancestral genomic DNA sequences using an ML approach (not on the basis of sequence reconstruction), a similar approach may not be tractable for protein sequences, given the significantly larger number of parameters to be estimated.
The method we introduce for inferring ancestral amino acid composition is based on the insight that the amino acid composition of conserved residues in present-day proteins, i.e., those residues which are unchanged between an ancestral sequence and any given descendant sequence, is determined by two factors: (1) the amino acid composition within ancestral sequences, and (2) the relative probability of conservation of each amino acid between an ancestral and an extant descendant sequence. Reversing this logic, given the amino acid composition of conserved residues and the relative probability of conservation of each amino acid, the amino acid composition within ancestral sequences can be inferred. Our approaches for identifying and estimating the composition of conserved residues, and estimating the relative probability of conservation of different amino acids are discussed subsequently (see Methods).
Simulations of sequence evolution have been employed previously to evaluate alternative methods for reconstructing events in the evolutionary past. For example, they have been used to assess the accuracy of methods for building phylogenetic trees (Saitou and Nei 1987) and reconstructing ancestral protein sequences (Zhang and Nei 1997 ). We introduce simulations here to model errors in the estimate of amino acid composition of ancestral sequences. Using these models, the bias in the estimated ancestral amino acid frequencies might be compensated for, allowing the estimates to be placed within a confidence interval.
Using our approach, we have estimated the amino acid composition of a set of 65 proteins within the LUA. We infer that within this set of proteins many of the amino acids believed to have been most abundant in the prebiotic environment (Miller 1953 , 1987 ; Kvenvolden et al. 1970 ), including glycine, alanine, aspartic acid, and valine, were used more frequently within proteins of the LUA than within those of modern species. On the other hand, amino acids believed to have been rare or unavailable prebiotically, including cysteine, tryptophan, tyrosine, and phenylalanine, were generally used much less frequently within proteins of the LUA. This may reflect the order of addition of these two groups of amino acids to the genetic code.
Methods
Basis of Approach
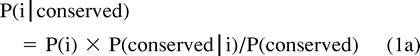
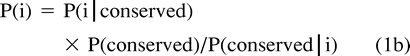
Estimating Probabilites
Identifying Conserved Residues Using MP Sequence Reconstruction
Let P′(i), P′(conserved), P′(conserved|i), and P′(i|conserved) denote the estimated values of P(i), P(conserved), P(conserved|i), and P(i|conserved), respectively. To estimate P′(conserved) and P′(i|conserved), conserved residues must first be identified. Although conserved residues are frequently defined as residues which are identical in all descendant sequences, this definition is too restrictive for our purposes. Instead we identify, as well as possible, all cases in which a residue remains unchanged between the ancestor and any of its descendant sequences; this is consistent with the definition of P(conserved) in equations (1a) and (1b) . To facilitate this process, ancestral sequences are partially reconstructed through MP (Eck and Dayhoff 1966 ), and these are compared with modern sequences to identify residues conserved between the ancestor and descendant (fig. 1B ). At each site s along the sequence, the number of identities between the inferred ancestor and each of the descendant sequences is tabulated to determine P′(conserved) at s. For example, if three of the four descendant sequences retain the original residue at site s, P′(conserved) at s is 0.75. P′(conserved) is then averaged over all sites. P′(i|conserved) simply represents the composition of sites identified as conserved between the ancestor and any of the descendants (fig. 1B ).
As illustrated in figure 1 , this method does not identify all instances in which a residue has been conserved between the ancestor and descendant, resulting in errors in P′(conserved) and P′(i|conserved). However, this method is preferable to counting as conserved only those sites identical in all descendants because on the basis of that criterion the actual conserved residues would be much more undercounted and thus P′(conserved) and P′(i|conserved) would be even more biased. Importantly, error in P′(i) arising through error in P′(conserved) and P′(i|conserved) may be modeled through simulation and taken into account in the derivation of the final estimates of P(i) (see Modeling Error in P′(i) Through Simulation below).
Estimating P(conserved|i) Using Subsitutition Probability Matrices
In theory, in addition to P′(conserved) and P′(i|conserved), P′(conserved|i) could also be obtained through a comparison of descendant sequences with partially reconstructed ancestral sequences. However, obtaining estimates of all three probabilities in such a manner would lead to values of P′(i) equivalent to the composition of the partially reconstructed sequences (once the values are normalized to sum to one); these P′(i) values are expected to be biased toward those amino acids which have a high relative probability of conservation (this expectation was confirmed through simulations; data not shown). To overcome this difficulty, substitution probability matrices based on empirical protein data are introduced as an independent means for obtaining P′(conserved|i).
A PAM 1 matrix represents the global average substitution probabilities that will result in approximately one substitution per 100 residues when applied to a sequence with the same average composition as the sequence set used to derive the matrices. Substitution probabilities for greater evolutionary distances can be computed as powers of the PAM 1 matrix (Dayhoff, Schwartz, and Orcutt 1978 , pp. 345–352); this requires the assumption that substitution probabilities are independent of time and position within the sequence. For modeling the composite evolution of a large sequence set in which only average substitution probabilities are required, this assumption is a reasonable first approximation, one which is frequently used in modeling protein evolution (Zhang and Nei 1997 ). Values for P(conserved|i) are then represented by the diagonal elements of the PAM probability matrix corresponding to a given evolutionary distance (Dayhoff, Schwartz, and Orcutt 1978 ). That is, P(conserved|i) is the probability that amino acid i is replaced by itself over the specified evolutionary distance.
Using this approach to estimate P(conserved|i) thus requires that a model of evolution based on a set of empirical matrices be assumed, which is common for the evolutionary analyses of proteins. Because a molecular clock is assumed, regardless of the evolutionary relationships between the descendant sequences, for any amino acid in the ancestor the expected fraction of descendant sites conserved is equal to the probability of conservation for that amino acid at a given evolutionary distance. As noted previously, error in the estimates of P(i) arising through deviation from the assumed molecular clock may be modeled and compensated for through simulation.
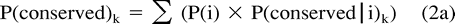
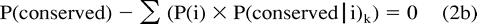
In our approach, although P(conserved) and P(i|conserved) may be estimated as described previously, P(i) and the distance k from which P(conserved|i) may be estimated are both unknown. We therefore developed the following method to identify the matrix distance separating ancestral and offspring sequences, and thus P(conserved|i) and P(i), given P(conserved) and P(i|conserved). PAM matrices of successively increasing distance k are used to supply P(conserved|i)k. The method empirically determines the distance k which best fits our estimates of P(conserved) and P(i|conserved) as follows:
(1) For each k (a) estimate P(i) by P(i)k = P(i|conserved) × P(conserved)/P(conserved|i)k and (b) because these probabilities should sum to 1.0, calculate the normalization factor αk such that αkΣ P(i)k = 1.0.
(2) Choose the matrix for which |log αk| is minimized, i.e., the matrix k for which the probability estimates of P(i) require the least amount of normalization.
The corresponding normalized values of P(i)k indicate P(i). It may be shown that our method finds the integer k whose estimates of P(conserved|i) and P(i) minimize the term given in equation (2c) (see Appendix). The same method may be used to derive P′(conserved|i) and P′(i), given P′(conserved) and P′(i|conserved).
Assuming that a representative subset of proteins within a genome is being analyzed, substitution matrices based on good global alignments of generic protein families are most likely to accurately reflect probabilities relating to the evolution of the set. Such matrices include the original PAM matrix of Dayhoff (Dayhoff, Schwartz, and Orcutt 1978 ) and its updated version by Jones, Taylor, and Thornton (1992) . Because the matrices of Gonnet, Cohen, and Benner (1992) are based upon alignments of highly divergent homologs, which are inherently difficult to align, for our purposes the matrices of Jones, Taylor, and Thornton (1992) are preferable. (These matrices are based on alignments of relatively closely related proteins [>85% identity].) The matrices of Jones, Taylor, and Thornton (1992) , based on release 22 of the SWISS-PROT databank, the current version of which is described in Bairoch and Apweiler (2000) , were therefore used for all the analyses described here. This series is frequently used to represent substitution probabilities for evolutionary modeling (see, for example, Yang, Kumar, and Nei 1995 ; Zhang and Nei 1997 ). It should be noted that neither matrices based on local alignments, such as BLOSUM, which are biased toward protein interiors (Henikoff and Henikoff 1992 ), nor matrices derived from specific protein subclasses, such as transmembrane proteins (e.g., Jones, Taylor, and Thornton 1994 ), are suitable to provide estimates of P(conserved|i).
Lineage-specific versus pooled estimates
Rather than pooling data from multiple species to arrive at P′(conserved), P′(i|conserved), P′(conserved|i), and ultimately P′(i), independent estimates of P(i) for each lineage can be made and then averaged. This allows the elimination of the assumption of a molecular clock. However, the differences in P′(i) arrived at using either pooled or lineage-specific data were insignificant (data not shown). Because pooling data from all lineages facilitates modeling and correction for error in the estimates, estimates based on pooled data were used.
Estimating Amino Acid Composition in the LUA
Choice of Protein Set
Although there has been much recent discussion of the nature of the LUA (see for example Woese 1998 ), it is sufficient for our purposes to think of the LUA as either a heterogeneous or a homogeneous population of organisms which eventually diverged into the three primary lineages. On the basis of their presence in eubacteria, archaea, and eukaryotes, approximately 325 proteins are proposed to have been present in the LUA (Kyrpides, Overbeek, and Ouzounis 1999 ).
Orthologous proteins (homologs which arose through speciation) that are present in a group of modern day species may be inferred to have been present in their last common ancestor. To assemble a set of proteins that were likely present in the LUA, we used the Clusters of Orthologous Groups (COG) database (Tatusov, Koonin, and Lipman 1997 ; Tatusov et al. 2001 ) to select all orthologous proteins common to a group of eight species which included members from each of the primary lineages: two archaea, Archaeoglobus fulgidus and Methanobacterium thermoautotrophicum; one eukaryote, Saccharomyces cerevisiae; and five eubacteria, Aquifex aeolicus, Thermotoga maritima, Synechocystis PCC6803, Escherichia coli, and Bacillus subtilis. These species were chosen because their phylogenetic diversity was expected to maximize the extent and accuracy of ancestral sequence reconstruction. Two hundred fifteen protein families in the COG database are present in all eight species.
Inclusion in the reconstruction of paralogs (homologs arising through gene duplication) which arose before the speciation event separating the species would invalidate the assumed species phylogenetic tree. In the COG database, both orthologs and closely related paralogs are grouped together into protein families. To minimize the possibility of including incorrect paralogs in sequence reconstructions, COG protein families were included either if they had only one protein member per species for all species other than yeast or if the existing paralogs could clearly be determined to have arisen after species divergence. (The tree provided with each COG protein family was used to resolve all issues regarding the protein phylogenetic tree.) For many protein families, yeast has a paralog of mitochondrial origin in addition to the true ortholog. The yeast homolog which clusters with the archaeal species was assumed to represent the true ortholog and was chosen for the analysis. This restriction reduced the protein set to 105 families.
Because lateral transfer also invalidates the assumed species tree, it is important to minimize the number of laterally transferred proteins included in the set. Whole genome analyses have suggested that species phylogenetic trees are broadly consistent with the small subunit rRNA (SSU rRNA) tree (and the trees of the majority of informational proteins, i.e., proteins playing a role in replication, transcription, or translation [Fitz-Gibbon and House 1999 ; Teichmann and Mitchison 1999] ). Therefore, we eliminated from our set all proteins whose trees were not consistent with the SSU rRNA tree (Olsen, Woese, and Overbeek 1994 ). For this purpose, we applied the criterion that the yeast and archaeal proteins should cluster on the protein tree. After this restriction was applied, 65 proteins remained in the set. These proteins are primarily classified as informational, most playing a role in translation. The COG protein families included in the analysis are listed in table 1 .
Application of Method
Multiple sequence alignments created by the program Clustal W (1.74) (Thompson, Higgins, and Gibson 1994 ) were input to an MP program, protpars, part of the phylogenetic software package PHYLIP (Felsenstein 1993 ). The phylogenetic tree based on ss rRNA sequence data (Olsen, Woese, and Overbeek 1994 ) was assumed for sequence reconstruction. On the basis of the comparison of the reconstructed and extant sequences, conserved residues were identified, allowing P′(i|conserved) and P′(conserved) to be derived. Using these estimates, P′(conserved|i) and P′(i) were determined as described previously. In total, 164,730 residues from the eight species were analyzed to arrive at P′(i). We refer to this estimate as P′(i)LUA. Table 2 shows the values of P′(i|conserved), P′(conserved), and P′(conserved|i) used to arrive at P′(i)LUA.
Modeling Error in P′(i) Through Simulation
Simulations are commonly employed to model sequence evolution; typically they are used to assess the performance of alternative phylogenetic methods on known data (see for example Zhang and Nei 1997 ). Instead, we employ simulations to model and correct for error in P′(i) so that confidence intervals may be determined for P(i)LUA. We anticipate error in P′(i|conserved), P′(conserved), and P′(conserved|i), and consequently in P′(i) because of the inability to identify all true conserved residues (fig. 1B ) and violation of the assumption of a molecular clock.
The simulations were performed as follows: a set of n ancestral sequences of determined length was randomly generated according to a given P(i). The resulting ancestral amino acid composition is referred to as P(i)S. The ancestral sequences were evolved to produce descendant sequences based on a phylogenetic tree with unequal branch lengths reflecting average evolutionary distances within the protein set and a substitution probability matrix representing PAM 1 (Jones, Taylor, and Thornton 1992 ). P′(i)S of the ancestral sequence set was derived using our method, and P(i)S − P′(i)S was determined. This difference is the error in P′(i)S.
The modeled sources of error were systematic and predictable. First, P′(conserved) is an underestimate of P(conserved) (values of 0.3425 and 0.4584, respectively). This underestimate can be explained by the fact that any sequence position in which the residue in at least one of the descendants has been conserved, but for which an ancestral residue was not assigned through MP, will lead to an undercount of P(conserved) (see fig. 1B ). The underestimate of P(conserved) causes P′(conserved|i)k to be chosen from a greater PAM distance k than would the true value of P(conserved) (the lower the conservation between sequences, the greater the evolutionary distance between them; see eq. (2a) ).
To understand how bias in P′(conserved) and P′(conserved|i) interact to result in bias in P′(i), it is useful to consider the ratio P′(conserved):P′(conserved|i) (see eq. (1b) ). With increasing PAM distance, the ratio P(conserved):P(conserved|i) for any amino acid i diverges from unity (fig. 2 ). This trend holds for PAM = 1 to 150, the range of distances relevant to our data set. For those amino acids for which P(conserved):P(conserved|i) < 1.0, such as tryptophan, the ratio decreases with increasing PAM, whereas for those amino acids for which P(conserved):P(conserved|i) > 1.0, such as alanine, the ratio increases (fig. 2 ). Therefore, as a consequence of the underestimate of P(conserved), it is observed that for those amino acids for which P(conserved):P(conserved|i) < 1.0, P′(conserved):P′(conserved|i) is an underestimate (P′(conserved):P′(conserved|i)/P(conserved):P(conserved|i) < 1.0; table 3 , columns 2 and 3; fig. 2 ). In contrast, for those amino acids for which P(conserved):P(conserved|i) > 1.0, this ratio is an overestimate (table 3 , columns 2 and 3; fig. 2 ).
On the other hand, P′(i|conserved) is generally an underestimate for those amino acids for which P(conserved):P(conserved|i) > 1.0 and an overestimate for those for which P(conserved):P(conserved|i) < 1.0 (table 3 , column 4), arginine being the only exception. These observed biases can be explained by the fact that those amino acids with a relatively high P(conserved|i) are more likely to be conserved in enough descendant sequences such that the ancestral residue can be assigned, and therefore, they are more likely to be correctly identified as conserved than those amino acids with a relatively low P(conserved|i). Thus, values for P′(i|conserved) are biased in favor of amino acids with a relatively high P(conserved|i) (table 3 , column 4).
Qualitatively, bias in P′(conserved):P′(conserved|i) and P′(i|conserved) counteract in equation (1b) . For those amino acids with a relatively low P(conserved|i), P′(i|conserved) is an underestimate, whereas P′(conserved):P′(conserved|i) is an overestimate (table 3 , columns 2–4). The reverse is true for those amino acids with a relatively high P(conserved|i). Quantitatively, on the other hand, bias in P′(i|conserved) and P′(conserved):P′(conserved|i) do not precisely offset, so there is net error in P′(i).
The predictable nature of the bias revealed in this way provided confidence that simulations could be used to model and correct for error in estimates based on real sequence data.
Placing a Confidence Interval on P(i) in the LUA
Simulations were performed with parameters chosen to model the LUA data set. To represent the ancestral sequence set, 65 proteins of 320 residues each (the average length of the analyzed sequences) were randomly generated using P′(i)LUA. To generate a phylogenetic tree representing the sequence set, a distance matrix containing average pairwise distances between the species for the entire LUA protein set was first determined using the program protdist of the PHYLIP package (Felsenstein 1993 ). The program Fitch of the PHYLIP package was used to convert these distances into an unrooted phylogenetic tree with branch lengths. This tree was converted into a tree with its root between the eubacterial lineage and the lineage giving rise to the archaea and eukaryotes. After simulated evolution to create eight descendant sequences, 166,400 residues (a number similar to the 164,730 residues of the real data set) were available to derive P′(i|conserved)S, P′(conserved)S, P′(conserved|i)S, and thereby P′(i)S. P(i)S − P′(i)S, or the error in P′(i)S, was then determined for each simulation. One hundred simulations were performed to derive statistics (mean and standard deviation) of the error in P′(i)S. This number of simulations was sufficient for the values of these statistics to stabilize.
The mean and standard deviation of error in P′(i)S over 100 simulations (table 4 , columns 4 and 5) were used to place P(i)LUA within 95% confidence intervals. First, biases in P′(i)LUA were corrected by adding to them the mean simulated error (table 4 , column 6). Lower and upper bounds of 95% confidence intervals for P(i)LUA were determined by subtracting or adding, respectively, twice the standard deviation of error in P′(i)S to these corrected values (table 4 , columns 7 and 8). These confidence intervals are based on the assumption that the simulations accurately model error in P′(i)LUA.
Summary of the Method Used for Estimating Composition of Protein Set in LUA
We have explained in detail previously in this article each step of our method and its application to infer the amino acid composition of a set of proteins in the LUA. It may be helpful to the reader if we now summarize it. First, the protein set was chosen. Next, the proteins were aligned and partial ancestral sequences inferred. Conserved sequence positions were then identified, allowing P(i|conserved) and P(conserved) to be estimated. P(conserved|i) was estimated using a substitution probability matrix. An initial estimate of P(i) could then be made. Finally, sequence simulations were used to model error in estimates of P(i), allowing a confidence interval to be placed on P(i) in the LUA.
Results
Inferred Change in Amino Acid Composition Since the LUA
Using the probabilistic approach and simulations described here, and assuming the Jones, Taylor, and Thornton (1992) series of matrices as a model of amino acid substitution probabilities, the amino acid composition of 65 proteins in the LUA was inferred. Figure 3 shows the change in frequency of individual amino acids within this protein set between the LUA and today (see also table 4 ). The modern composition upon which this comparison is based represents the average composition of this protein set in the eight modern species examined. Nine amino acids, alanine, asparagine, aspartatic acid, glycine, histidine, isoleucine, serine, theonine, and valine, are inferred to have occurred more frequently in this protein set in the LUA than today (fig. 3A ). On the other hand, eight amino acids, cysteine, glutamine, glutamic acid, leucine, lysine, phenylalanine, tryptophan, and tyrosine, are inferred to have been used less frequently in the LUA (fig. 3B ). The confidence intervals on P(arginine), P(methionine), and P(proline) (fig. 3C ) do not allow an inference to be made as to whether or how these amino acids have changed in frequency since the LUA.
Discussion
Inferred Order of Entry of Amino Acids into the Genetic Code
On the basis of the change in frequency of amino acids between the LUA and today, we may make inferences regarding the establishment of the genetic code (Brooks and Fresco 2002 ). It is reasonable to assume that as the genetic code evolved, newly assigned amino acids adopted codons used infrequently in coding sequences to minimize the structural disruption of the encoded protein (Osawa et al. 1992 ). Consequently, new amino acids would have been introduced gradually into existing primitive proteins. Thus, at the time the genetic code became fully established, those amino acids which had been added relatively early would have been overrepresented and those which had been added relatively late would have been underrepresented, relative to the composition of modern proteins. Starting from such early biased amino acid composition, primitive proteins would have proceeded to evolve toward their modern-day compositions. In such a scenario, the amino acids that were introduced into the genetic code relatively early should have decreased in frequency over the course of evolution, whereas those amino acids added relatively late should have increased in frequency (i.e., between the establishment of the genetic code, the LUA, and today).
The nine amino acids which have decreased in frequency between the LUA and today (fig. 3A ) may thus be inferred to have been introduced into the code early. Most of these amino acids are among those presumed to have been most abundant in the prebiotic environment, as inferred through spark tube simulations (Miller 1953 , 1987 ) and analysis of the Murchison meteorite (Kvenvolden et al. 1970 ). In contrast, the eight amino acids which have increased in frequency between the LUA and today (fig. 3B ), and which are thus inferred to have been late additions to the code, include several of the most biosynthetically complex amino acids (for example, all three aromatic amino acids, which share a common, complex metabolic intermediate, are inferred to have been late additions); most of these are presumed either to have been nonexistent or of very low abundance in the prebiotic environment. Two of these, cysteine and tryptophan, are both conservatively estimated to have been less than half as frequent within this protein set in the LUA than today.
We emphasize that the validity of the inferences drawn in this study depend upon the reliability of the Jones, Taylor, and Thornton (1992) substitution probabilities for modeling evolution over very long time periods and along all lineages. With the development of lineage-specific models of evolution, estimates of ancestral amino acid composition can be expected to improve. In the meantime, although there are undoubtedly limitations to using the matrices of Jones, Taylor, and Thornton (1992) to model evolution since the LUA, we feel they provide the best available estimates of these substitution probabilities. It is noteworthy that previously, on the basis of an independent approach, cysteine, tyrosine, and phenylalanine were inferred to have been used less frequently in proteins of the LUA than today (Brooks and Fresco 2002 ).
The inferences drawn here regarding the relatively early or late introduction of amino acids into the genetic code are generally consistent with earlier proposals which were based on the presumed presence or absence of various amino acids in the primordial environment (see for example Wong 1975 ). However, for a few amino acids our assignment as early or late is not in keeping with earlier ideas. For example, histidine and asparagine, believed to have been absent in the prebiotic environment, are both inferred through the present work to have been added to the code relatively early, whereas glutamate, believed to have been present in the prebiotic environment, is inferred to have been added late (figs. 3 and 4 ). Interestingly, each of these three amino acids share a block of four codons with a second amino acid: histidine with glutamine, asparagine with lysine, and glutamate with aspartate (fig. 4 ). Codon capture, in which one amino acid loses some of its codons to another, is commonly proposed as a mechanism for introducing amino acids, especially later arriving ones, into the code (Crick 1968 ; Wong 1975 ). Consistent with codon capture, it is plausible that one amino acid was added to the four-codon block first and that this amino acid later gave up two of its codons to the second amino acid which now shares the block.
Accordingly, those amino acids which were originally assigned to the codon block (i.e., aspartate, asparagine, and histidine) would have the appearance of being added to the code early, whereas those which were added to the block later through codon capture (i.e., glutamate, lysine, and glutamine) would have the appearance of being added late. Therefore, early and late amino acids do not correspond to a strict chronological order of introduction into the code. Instead, as defined here on the basis of the changing amino acid frequencies, early amino acids are probably those which at some point lost some of their codons through codon capture and consequently became less frequent over time within proteins, whereas late amino acids are those which entered the code through codon capture, did not subsequently lose any of their codons, and therefore became more frequent over time (fig. 4 ). Finally, it is worth noting that the distinction between amino acids inferred here to have been added to the genetic code early or late does not at all correlate with the two main structural classes of the aminoacyl-tRNA synthetases. This is consistent with the earlier suggestion that these enzymes probably had no specific role in the evolution of the genetic code (Woese 2000 ).
Existing ideas regarding the origin and evolution of the genetic code have been based largely on theoretical investigations and experiments involving oligonucleotide aptamer binding of amino acids (reviewed in Knight, Freeland, and Landweber 1999 ). The present findings suggest that notwithstanding the impact of mutation over the long course of molecular evolution, with the aid of the appropriate analytical tools and insights, the sequences of contemporary proteins also provide an important avenue for exploring these early evolutionary events.
Appendix
Given P(i) and P(conserved), one may find the matrix of distance k to minimize (2c).

Thus, by choosing the value k which minimizes |log αk|, we are finding the PAM distance k whose corresponding estimates of P(i) and P(conserved|i) minimize (2c).
William Taylor, Reviewing Editor
Abbreviations: MP, maximum parsimony; ML, maximum likelihood; LUA, Last Universal Ancestor; COG, Clusters of Orthologous Groups.
Keywords: amino acid composition protein evolution Last Universal Ancestor substitution probability matrices genetic code
Address for correspondence and reprints: Mona Singh, Department of Computer Science, Princeton University, Princeton, New Jersey 08544. mona@cs.princeton.edu
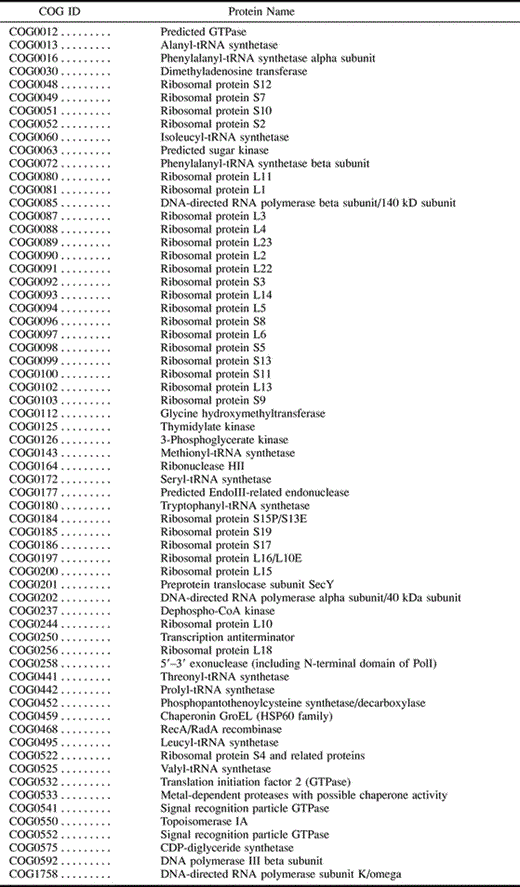
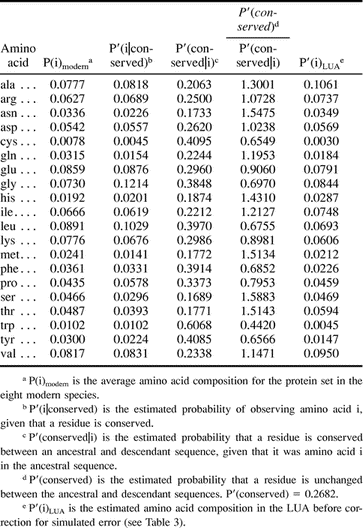
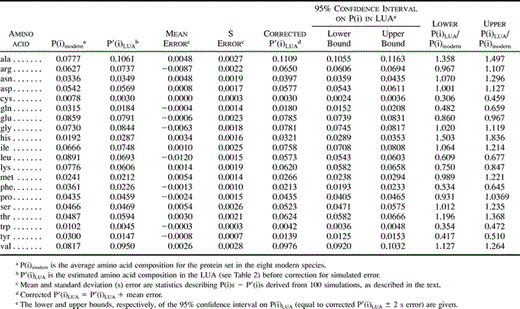
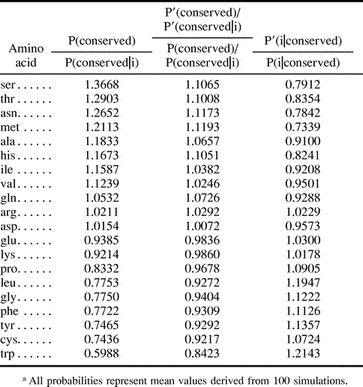

Fig. 1.—A, Illustration of the relationship between P(i), P(i|conserved), P(conserved), and P(conserved|i). A simplified schematic showing a section of a true ancestral sequence aligned with its modern descendants. Residues conserved between the ancestor and each descendant are underlined. P(i|conserved), P(conserved), and P(conserved|i) are based on so-identified conserved sequence positions. One can see from this example that the relationship expressed in equation (1b) holds. B, Estimation of P(conserved) and P(i|conserved). For estimation of P(conserved) and P(i|conserved), conserved residues are defined as those residues in the modern and inferred ancestral sequences which are identical. (Note that the ancestral sequence is inferred through parsimony and that residues do not have to be identical in all modern sequences to be defined as conserved.) Residues identified as conserved in this manner are underlined. P′(conserved) is the fraction of residues in all the descendant sequences which are identified as conserved, whereas P′(i|conserved) is the fraction of all conserved sites containing residue i. Assuming that the fourth residue in the true ancestral sequence is G, P′(conserved) and P′(G|conserved) are both underestimates, whereas P′(W|conserved), P′(P|conserved), and P′(C|conserved) are overestimates (compare with values in A)
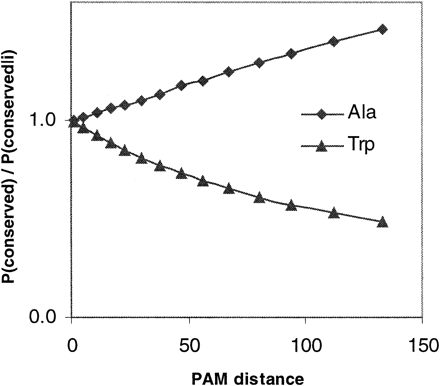
Fig. 2.—P(conserved)/P(conserved|i) diverges from unity with increasing PAM distance. The change in the ratio P(conserved)/P(conserved|i) with change in PAM distance is shown for two representative amino acids: one, alanine, for which P(conserved)/P(conserved|i) > 1.0, and the other, tryptophan, for which P(conserved)/P(conserved|i) < 1.0. The same trend is observed for all other amino acids
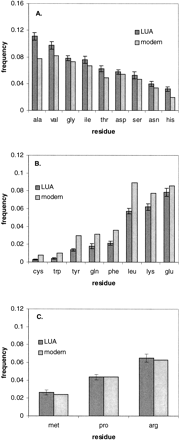
Fig. 3.—Change in amino acid frequencies between the LUA and today. The 95% confidence intervals on frequencies in the LUA given in table 3 are represented here as error bars. “Modern” reflects the average amino acid composition for the protein set in the eight modern species. Amino acids with inferred (A) decrease, (B) increase, or (C) no change in frequency between the LUA and today
Fig. 4.—Early or late addition to the genetic code versus presence in the prebiotic environment. Amino acids and their corresponding codons are coded as follows: underlined = early; outline = late; normal = undetermined; and gray highlight = most abundant prebiotic. Assignment of amino acids as early, late, or undetermined is based on this work. Assignment of amino acids as most abundant prebiotic is based on Miller (1987)
We are grateful to Warren Ewens for providing advice on the statistical treatment of error in the estimate of P(i) and for comments on the manuscript. We also wish to thank Nick Goldman, Robert Osada and Carl Kingsford for thoughtful suggestions on the manuscript. D.J.B. was supported by predoctoral traineeships from NIH grant 2T32GM07388-22 and NSF grant DGE 9972930, A.M.L. is supported by the Wellcome Trust, and M.S. is supported by NSF Pecase Grant MCB-0093399. The computational facility utilized for this work was obtained with funds provided to J.R.F. by the Department of Defense through MEDCOM at Fort Detrick, MD.
References
Bairoch A., R. Apweiler,
Brooks D. J., J. R. Fresco,
Dayhoff M. O., R. M. Schwartz, B. C. Orcutt,
Eck R. V., M. O. Dayhoff,
Felsenstein J.,
Fitz-Gibbon S. T., C. H. House,
Galtier N., N. Tourasse, M. Gouy,
Gerstein M.,
———.
———.
Gonnet G. H., M. A. Cohen, S. A. Benner,
Henikoff S., J. Henikoff,
Jones D. T., W. M. Taylor, J. M. Thornton,
Knight R. D., S. J. Freeland, L. F. Landweber,
Kvenvolden K., J. Lawless, E. Pering, E. Peterson, J. Flores, C. Ponnamperuma, I. R. Kaplan, C. Moore,
Kyrpides N., R. Overbeek, C. Ouzounis,
Miller S. L.,
———.
Olsen G. J., C. R. Woese, R. Overbeek,
Osawa S., T. H. Jukes, K. Watanabe, A. Muto,
Saitou N., M. Nei,
Tatusov R. L., E. V. Koonin, D. J. Lipman,
Tatusov R. L., D. A. Natale, I. V. Garkavtsev, T. A. Tatusova, U. T. Shankavaram, B. S. Rao, B. Kiryutin, M. Y. Galperin, N. D. Fedorova, E. V. Koonin,
Teichmann S. A., G. Mitchison,
Thompson J. D., D. G. Higgins, T. J. Gibson,
———.
Wolf Y. I., N. V. Grishin, E. V. Koonin,
Yang Z., S. Kumar, M. Nei,

{kind=link}
{kind=link}
{kind=link}
{kind=link}