





Abstract
To prevent progressive telomere shortening as a result of conventional DNA replication, new telomeric DNA must be added onto the chromosome end. The de novo DNA synthesis involves elongation of the G-rich strand of the telomere by telomerase. In human cells, the CST complex (CTC1-STN1-TEN1) also functions in telomere replication. CST first aids in duplication of the telomeric dsDNA. Then after telomerase has extended the G-rich strand, CST facilitates fill-in synthesis of the complementary C-strand. Here, we analyze telomere structure after disruption of human CTC1 and demonstrate that functional CST is essential for telomere length maintenance due to its role in mediating C-strand fill-in. Removal of CTC1 results in elongation of the 3΄ overhang on the G-rich strand. This leads to accumulation of RPA and telomeric DNA damage signaling. G-overhang length increases with time after CTC1 disruption and at early times net G-strand growth is apparent, indicating telomerase-mediated G-strand extension. In contrast, C-strand length decreases continuously, indicating a deficiency in C-strand fill-in synthesis. The lack of C-strand maintenance leads to gradual shortening of the telomeric dsDNA, similar to that observed in cells lacking telomerase. Thus, telomerase-mediated G-strand extension and CST-mediated C-strand fill-in are equally important for telomere length maintenance.
INTRODUCTION
Mammalian telomeres exist as nucleoprotein complexes composed of a six protein complex called shelterin and kilobases of repeated sequence dsDNA. The dsDNA is made up of TTAGGG·AATCCC repeats and it terminates in a 12–400 nt 3΄ G-rich overhang (1,2). This overhang (termed the G-overhang) serves as a substrate for the enzyme telomerase which elongates the overhang by adding TTAGGG repeats to the 3΄ terminus. A portion of the elongated overhang is subsequently converted to dsDNA by DNA polymerase. Thus, the combined action of telomerase and DNA polymerase are needed to compensate for the telomere shortening that occurs due to the inability of DNA polymerase to completely replicate the DNA 5΄ end. Shelterin prevents the DNA terminus from triggering ATM/ATR-mediated DNA damage signals and facilitates telomere replication (3–5). The shelterin components TRF1 and TRF2 bind the dsDNA while POT1 binds the G-overhang. TPP1 dimerizes with POT1 and links POT1 to TRF1/2 via TIN2.
Telomere replication is a multi-step process that involves not only the conventional replication machinery, telomerase and shelterin, but also the CST complex (CTC1-STN1-TEN1) and various other accessory factors. In human cells, the telomeric dsDNA is replicated by the conventional replication machinery aided by TRF1, CST and multiple helicases (5,6). G-overhangs are generated on the telomere replicated by lagging strand synthesis through removal of the RNA primer and sub-terminal placement of the final Okazaki fragment (7). Overhangs are generated on the telomere replicated by leading strand synthesis through nuclease resection (7,8). TPP1 then stabilizes telomerase association with the telomeres, allowing telomerase to extend the G-strands by ∼60 nt (4,9–11). Telomerase extension occurs shortly after duplex replication and G-overhang generation. However, maturation of the G-overhang to its final length does not occur until late S/G2 when complementary C-strand DNA is synthesized by a DNA polymerase (most likely polymerase α-primase) in a process called C-strand fill-in (9). Given that the replisome is unlikely to persist at this time, it has been unclear how DNA polymerase is brought to the G-overhang for the C-strand fill-in reaction. We now demonstrate that human CST is essential for the C-strand fill-in reaction and hence for telomere length maintenance in human cells.
Mammalian CST is a trimeric complex that has structural similarity to RPA (Replication Protein A) and like RPA, appears to bind ssDNA via multiple OB-folds (12–16). However, RPA and CST exhibit significant differences in how individual subunits contribute to DNA binding and the overall architecture of the resulting DNA-protein complex (13,14). Also, CST binds preferentially to telomeric G-strand DNA in a length dependent manner whereas RPA does not (13,17). Importantly, the roles of CST and RPA appear to be quite distinct with CST function restricted to the resolution of specific problems associated with DNA replication.
One role of human CST is to aid in replication through the repetitive telomeric dsDNA (6,18). Depletion of STN1 or TEN1 slows replication through this region and results in periodic telomere loss and/or a fragile telomere phenotype in cells with long telomeres (6,18–20). CST also appears to participate in several aspects of G-overhang maturation. First the complex seems to limit G-strand extension by telomerase (17). Second, CST action is needed for DNA polymerase to initiate the C-strand fill-in reaction (6,19). CST depletion leads to a delay in C-strand synthesis which results in the maintenance of extended G-overhangs throughout G2 of the cell cycle (6).
CST also has less well understood genome wide roles in the resolution of replication stress (21,22). STN1 localizes to GC-rich repetitive sequences after hydroxyurea-induced replication fork stalling and STN1 depletion leads to DNA breakage at these sites (22). STN1 or TEN1 depletion also results in anaphase bridges and STN1 depletion causes a decrease in firing of dormant or late replication origins (18,21). The mechanism whereby CST facilitates resolution of replication issues at telomeres and elsewhere in the genome remains unclear. However, recent studies suggest that CST may act by directing loading or unloading of partner proteins, such as DNA polymerase or Rad51, on DNA in a manner akin to how RPA directs the formation and dissolution of complexes required for DNA replication, repair and recombination (13,22–25).
CST is essential for human health as mutations in CTC1 or STN1 cause the disease Coats plus and symptoms of dyskeratosis congenita (26–29). Patients have biallelic mutations that usually lead to complete inactivation of one allele and generation of mutant protein from the second allele. Coats plus patients present with retinal telangiectasia, intracranial calcifications, osteopenia, and gastrointestinal bleeding. Dyskeratosis congenita is a telomere maintenance disorder where telomere shortening limits cell proliferation and patients ultimately die of bone marrow failure. However, some Coats patients display normal telomere length, suggesting that both telomeric and non-telomeric deficiencies underlie the disease (27–29).
Mice with complete CTC1 deletion experience bone marrow failure and die within two months of birth (30). They also display more severe telomere defects than Coats plus patients, including extensive telomere loss and end-to-end fusion of chromosomes. The lesser telomere deficiency seen in patients may partially reflect the hypormorphic nature of the CTC1 and STN1 mutations. However, differences in mouse and human telomere biology may also come into play as some shelterin components and aspects of telomere regulation differ between the two species (8,31–33). To address the effects of complete CTC1 loss in humans, we have generated HCT116 cells with a conditional CTC1 gene disruption. Analysis of these cells indicates that the effects of CTC1 removal do in fact differ between human and mouse cells. Moreover, our work uncovers the essential nature of CST-mediated C-strand fill-in for telomere length maintenance and demonstrates how a balance between CST and POT1 activity at the G-overhang is needed to prevent the telomere from triggering a DNA damage response.
MATERIALS AND METHODS
Cell culture and generation of CTC1F/F cells
293-AAV and 293T cells were grown in DMEM and HCT116 cells in McCoy's medium supplemented with 10% FBS, antibiotics, and glutamine. The CTC1 targeting construct (CTC1-3loxP), contained a fragment of the CTC1 genomic locus encompassing exons three through six. A loxP site was engineered into intron four and a neo cassette flanked by Flp recombinase and loxP sites was inserted into intron five. Introns 4 and 5 were chosen for LoxP insertion because introns 1–3 contained repetitive sequence and hence were unsuitable as sites for targeted integration. The construct was subcloned into pAAV MCS (pAAV-CTC1-3loxP, Figure 1A) and co-
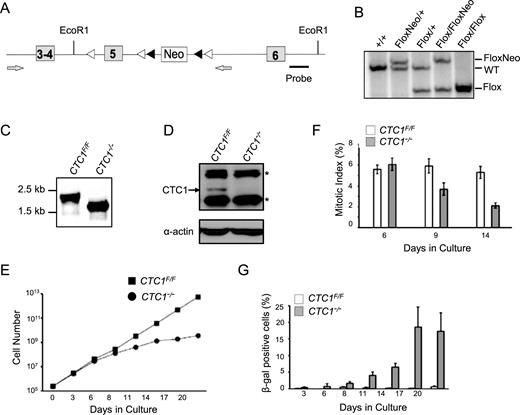
CTC1 gene disruption in HCT116 cells. (A) Modified CTC1 gene locus. Exon 5 was flanked by loxP sites (white arrowheads) and the neo marker by FRT sites (black arrowheads). Neo was removed by FLP recombinase after targeting of each allele. Grey arrows: primers used to verify loss of Neo and exon 5. (B) Southern blot showing modification of CTC1 gene locus. EcoR1 digested genomic DNA was hybridized with probe shown in (A). (C) PCR to verify gene disruption with primers flanking exon 5. CTC1 conditional cells (CTC1F/F cells expressing Cre-ER) were grown with tamoxifen (CTC1−/−) or without tamoxifen (CTC1F/F) for 3 days. (D) Western blot showing loss of CTC1 from CTC1−/− cells. CTC1 conditional cells were grown with/without tamoxifen for 7 days. Blot was probed with antibody to CTC1 or actin as a loading control. Arrow: CTC1, *: cross-reacting bands. (E and G) CTC1 conditional cells grown with/without tamoxifen for the indicated number of days. (E) Representative growth curves showing decreased proliferation after CTC1 disruption. (F) Quantification of mitotic index based on staining with phospho-H3 (mean ± S.E.M., n = 3 independent experiments). (G) Quantification of β-galactosidase positive cells (mean ± S.E.M., n = 3 independent experiments).
transfected with pAAV-RC and pAAV-helper into 293-AAV cells to make adeno-associated virus (AAV) (34). HCT116 cells were infected with AAV particles, cultured for 48 hrs then diluted to single cells and selected with G418 (0.5 mg/ml). Clones were screened by PCR for integration at the CTC1 locus using 5΄-CTTAGTCCATGTTCCCTGCAACC and 5΄-TGGACGTAAACTCCTCTTCAGAC to monitor the 5΄ end of the integration site and 5΄-TTCTATCGCCTTCTTGACGAG and 5΄-TGCTGCAGAAAAGGAGCCTATG for the 3΄ end. One clone with correct integration at a single allele was then transfected with pCAGGS-FLPE-puro (Addgene #20733), selected with 1 μg/ml puromycin for 24 hrs and subcloned. Removal of the Neo cassette was verified by sensitivity to G418 and PCR using 5΄-AGCGCTGTTGGGAGAAGATTC and 5΄-GACTGATGTCAAGGGAAGTATGAC. This clone, referred to as CTC1F/+, was re-infected with AAV and subject to G418 selection and FLP-mediated removal of Neo to obtain CTC1F/F cells. To introduce Cre recombinase, retrovirus was generated by co-transfecting 293T cells with CreERT2-puro (Addgene, 22776), gag-pol, and env. Viral supernatant was used to infect HCT116 CTC1F/+ or CTC1F/F cells. Cells were subcloned by limiting dilution and selected with puromycin. Tamoxifen (Sigma, H7904) was added to 10 nM to induce Cre activity. CTC1 disruption was verified by PCR with primers 5΄-CTTAGTCCATGTTCCCTGCAACC and 5΄-CACAGTAAGGCCCTATTTCTAC. The gene disruption creates a premature stop codon within exon six and the resulting mRNA is a predicted target of nonsense mediated decay. If truncated protein is made, it would terminate at amino acid 217 which is at the start of the first OB fold. To overexpress POT1, CTC1F/F cells were transfected with pMIT vector encoding FLAG-POT1 and cells were selected by flow cytometry for Thy1 expression.
Southern hybridization to screen for correct gene targeting
Genomic DNA was isolated using Illustra Genomic prep (GE), digested with EcoRI, separated in 0.75% agarose gels and transferred to nylon membrane. Clones with correct CTC1 gene targeting (see Figure 1B) were identified by hybridization using the probe indicated in Figure 1A. The probe was generated by PCR with primers 5΄-AGGATGTGGGGGAAGGATGG and 5΄- GACAAAAAGGGAAATCACCTGAGC.
Growth curves and β-galactosidase assays
Growth curves were performed as previously described, with three repetitions for CTC1F/F cells, two for CTC1F/+ cells (20). For β-galactosidase staining, 5 × 105 cells at the indicated time points were plated in a 35 mm dish and stained according to the manufacturer's protocol (Cell Signaling, 9860).
G-overhang and telomere length analysis
Genomic DNA was isolated by proteinase K digestion and isopropyl alcohol precipitation then digested overnight with HinfI, MspI and RsaI. For G-overhang analysis, control samples were treated with ExoI for 48 h prior to restriction digestion. DNA was separated briefly in 1% agarose gels, the gels were dried and hybridized with (TA2C3)3 probe under non-denaturing conditions. The DNA was then denatured, the gels re-hybridized with the same probe and bands quantified by PhosphorImager. To control for differences in loading, band intensities from the native gel were normalized to the corresponding band from the denatured gel. To assess changes in overhang abundance rather than internal ssDNA, the normalized band intensity of the Exo1 digested sample was subtracted from the mock digested counterpart. For telomere length analysis, the DNA was separated in 0.7% agarose gels, denatured and either subject to in-gel hybridization with (TA2C3)3 probe or transferred to nylon membrane prior to hybridization with the same probe.
Telomere FISH and γ-H2AX staining
FISH was performed on MeOH/acetic acid prepared metaphase spreads as previously described (18), except samples were hybridized with CENPB-Cy3 pan-centromere PNA probe (5΄ATTCGTTGGAAACGGGA, Biosynthesis) in addition to TelC-Alexa488 PNA G-strand probe (5΄ CCCTAACCCTAACCCTAA, Biosynthesis) or TelG-Cy3 PNA C-strand probe (5΄ GGGTTAGGGTTAGGGTTA, Biosynthesis). Images were taken at a constant exposure time. For quantitative measurement of telomere length (qFISH), telomere fluorescence intensity was integrated using the TFL-TELO program (35). Signal free ends (SFE) and telomere fusions are quantified by eye.
For combined γ-H2AX, RPA and telomere staining, cells were synchronized with colchicine (0.1 ng/μl) for 2 h and then collected for staining. The cells were first swelled with hypotonic buffer (0.2% KCl and 0.2% tri-sodium citrate) and deposited on slides by cytospin (36). Cells were then fixed with 4% formaldehyde in PBS and followed by permeablization in KCM buffer (120 mM KCl, 20 mM NaCl, 10 mM Tris pH 7.5, 0.1% Triton). Slides were blocked in antibody-dilution buffer (20 mM Tris pH 7.5, 2% BSA, 0.2% fish gelatin, 150 mM NaCl, 0.1% Triton, 100 μg/ml RNase A) at 37°C for 15 mins. For staining, slides were stained with γH2AX (Millipore, 05-636) or RPA (Millipore, NA-18) in antibody dilution buffer overnight at 4°C in a humidified chamber. FISH was performed as previously described (36). γH2AX foci were scored on ∼100 chromosomes for each experiment. For combined FISH and whole cell staining of POT1-FLAG, cells were pre-extracted with permeabilization solution (0.5% Triton, 20 mM HEPES, 50 mM NaCl, 3 mM MgCl2 and 300 mM sucrose) for 5 min prior to formaldehyde fixation. Cells were stained with mouse anti-FLAG antibody (Sigma, F1804) and TelC-Alexa488 PNA G-strand probe as previously described (37).
Chromatin Immunoprecipitation
Cells were fixed with 1% formaldehyde for 20 min. then treated with 200 mM glycine for 10 min to quench the reaction. Cell were pelleted by centrifugation, suspended in swelling buffer (25 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.25% Triton X-100 and protease inhibitors) on ice for 10 min, pelleted and incubated in sonication buffer (50 mM HEPES, pH 7.9, 150 mM NaCl, 1 mM EDTA, 0.1% Sodium deoxycholate, 0.1% SDS, 1% Triton X-100 and protease inhibitors) and sonicated for 20 min. in a Bioruptor sonication system (Diagenode). Samples were centrifuged at 14 000 rpm for 10 min and the supernatant used for ChIP. Samples containing supernatant (0.3 mg protein), antibody (2 μg TRF2, Millipore, 05-521; 10 μg RPA, Millipore, NA-19; 10 μg γH2AX, Millipore, 05-636) and 20 μg bacterial DNA were incubated overnight at 4°C. Protein A/G PLUS agarose beads (Santa Cruz) were then added and samples incubated for 1 h at 4°C. Beads were washed sequentially with wash buffer A (20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 150 mM NaCl), buffer B (20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 500 mM NaCl), buffer C (20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 0.5% NP40, 0.5% sodium deoxycholate, 250 mM LiCl) and TE buffer (10 mM Tris–HCl, pH 8.0, 1 mM EDTA). The immunoprecipitate was eluted in 450 μl elution buffer (1% SDS, 0.1 M NaHCO3), and cross-linking was reversed by incubation at 65°C overnight. The eluate was brought to 10 mM EDTA, 40 mM Tris–HCl, pH 6.8 and treated with RNase A at 37°C for 1 h and protease K at 55°C for another hour and the DNA purified by phenol–chloroform extraction. The input and precipitated DNAs were analyzed by slot blot hybridization with (TA2C3)3 probe and the telomeric DNA signal quantified by Phosphorimager. The background from the no antibody control was subtracted and the amount of precipitated DNA was calculated as a percentage of the corresponding input.
Western blotting
Cells were lysed in NP-40 lysis buffer (20 mM Tris pH8.0, 100 mM NaCl, 1 mM MgCl2 and 0.1% NP-40) and 50 μg protein was separated by SDS-PAGE and transferred to nitrocellulose. The membrane was blocked with 5% milk and incubated with antibody to actinin (Santa Cruz), CTC1 (Millipore MABE1103, clone C482), TEN1 (20) and STN1 (21). After incubation with HRP-conjugated secondary antibody, blots were developed using Western Lightning ECL (Perkin Elmer) for actinin and STN1 or Supersignal ECL (Thermo) for TEN1. For analysis of POT1 expression, cell extracts were prepared as described (38) and POT1 was detected on Western blots with antibody to FLAG (Sigma) or POT1 (AbCam 124784).
Statistical methods
Data were analyzed by two-tailed Student's t test. P-values: *P < 0.05, **P < 0.01.
RESULTS
Conditional disruption of human CTC1
To address the role of CTC1 in human cells, we generated a conditional CTC1 gene disruption in HCT116 colon carcinoma cells by introducing loxP sites into the introns flanking exon 5 of the endogenous CTC1 locus (Figure 1A). Gene targeting was performed using adeno-associated virus (AAV) and clones were selected for neomycin resistance after each gene-targeting step. The marker cassette was removed from the resulting CTC1F/+ and CTC1F/F cells by expression of FLP recombinase (Figure 1B). Cre recombinase was then introduced by infection with retrovirus encoding a Cre-ER fusion protein. Individual clones were treated with tamoxifen to activate Cre-induced recombination and analyzed to verify deletion of exon 5. Several clones were isolated that exhibited efficient gene disruption and loss of CTC1 following tamoxifen treatment and one was chosen for further analysis (Figure 1C and D).
In initial experiments, we examined the effect of CTC1 loss on growth rate and cell cycle progression. Following tamoxifen addition, the growth rate remained unchanged for 4–6 days but then slowed substantially (Figure 1E, Supplementary Figure S1A). FACS analysis of DNA content and staining of mitotic cells with antibody to phosphorylated histone H3 indicated that the decrease in growth was accompanied by a decline in the fraction of cells in S and M phase and an increase in the sub G1, G2 and >4N populations (Figure 1F, Supplementary Figure S1C). There was a concomitant increase in both cell death and in the fraction of senescent cells exhibiting β-galactosidase staining (Figure 1G, Supplementary Figure S1D). Since depletion of CTC1 has been found to decrease the stability of STN1 and TEN1 in some cell types (20,39), we also examined whether CTC1 disruption in HCT116 cells affects the level of STN1 or TEN1. Western blot analysis revealed some decline in STN1 abundance but no obvious change in TEN1 (Supplementary Figure S1B).
Removal of human CTC1 causes modest telomere loss but telomere fusions are rare
In mouse, deletion of CTC1 causes rapid telomere loss, accumulation of γH2AX at the resulting unprotected ends, and an accompanying increase in chromosome fusions that lack detectable telomeric DNA at the fusion junction (39). To determine if deletion of human CTC1 also results in telomere loss and chromosome fusions, we used fluorescent in situ hybridization (FISH) to examine metaphase spreads from the CTC1 conditional HCT116 cells. Spreads were prepared from cells grown with or without tamoxifen for 0–14 days and then hybridized with both a PNA probe to the telomeric G-strand and a pan centromere probe (Figure 2A). The centromere probe was used to aid in the identification of fused, di-centric chromosomes. Interestingly, the FISH revealed only a low level of chromosome fusions (0.3% of total chromosomes exhibited fusions at day 14, Supplementary Figure S2A). Chromosomes that lacked staining at one or more telomeres were more apparent (Figure 2A, Supplementary Figure S2A) with the frequency of these signal free ends (SFE) increasing from ∼2% at the time of tamoxifen addition to ∼6% after 14 days of treatment. Over the same time period there was also a ∼2-fold increase in the fraction of chromosomes exhibiting sister telomere association (Supplementary Figure S2A) but again the overall level was quite modest (∼2%). Although, CST is known to cause problems with telomere duplex replication (6,18,20,39), the characteristic multiple telomeric signals (MTS) were not visible on individual chromatids. This may be because telomeres from HCT116 cells are relatively short (∼6 kb, see below) (6).
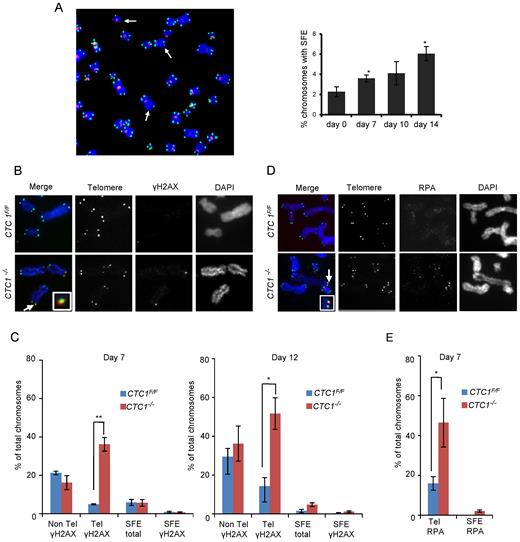
CTC1 disruption causes telomeric DNA damage signaling at telomeres that retain telomeric sequence. CTC1 conditional cells were grown with/without tamoxifen for the indicated times. (A) Left: Metaphase spread from cells grown with tamoxifen for 10 days. White arrows indicate partial telomere loss or full telomere loss (signal free ends, SFE). Chromosomes were hybridized with (C3TA2)3 telomere probe (green) and centromere probe (red) and stained with DAPI (blue). Right: Quantification of chromosomes showing one or more SFEs at the indicated time points, n = 3 independent experiments, mean ± S.E.M, ≥2000 chromosomes analyzed per time point. (B–E) Metaphase spreads showing γH2AX or RPA localization. Telomeres detected by FISH (green), γH2AX or RPA by immunostaining (red), chromosomes were counterstained with DAPI (blue). n = 3 independent experiments, mean ± S.E.M. ≥300 chromosomes scored per time point. (B) Images showing γH2AX staining at day 12. Arrows indicate co-localization, inserts show enlargement of same region. (C) Quantification of chromosomes with γH2AX foci on chromosome arms (Non-Tel), at one or more telomeres with detectable telomeric DNA (Tel) or at signal free ends (SFE). (D) Images showing RPA staining at day 7. (E) Quantification of chromosomes with RPA foci at telomeres.
DNA damage signaling is activated at telomeres that retain telomeric DNA
Since CTC1 deletion results in an increase in telomeres lacking detectable telomeric DNA, we asked whether these chromosome ends elicit a DNA damage signal and hence may be responsible for the observed growth inhibition. To detect damage signaling, we prepared metaphase spreads by cytospin at various times after CTC1 deletion and looked for γH2AX localization at the chromosome termini. γH2AX was detected by immunolocalization and telomeric DNA visualized by FISH (Figure 2B and C and Supplementary Figure S2B and C). The γH2AX staining revealed an increase in telomeric damage signaling that was apparent by day 4 and became more pronounced at days 7 and 12. However, the increase in telomeric γH2AX was mostly at chromosome ends that retained telomeric FISH signals rather than at the signal free ends. The proportion of signal free ends that lacked γH2AX was >65% and was similar for CTC1−/− and CTC1F/F cells. Thus, it appears that the signal free ends in CTC1−/− cells retain sufficient telomeric DNA to prevent activation of a damage response. This finding explains the lack of telomere fusions after CTC1 deletion.
The DNA damage signaling by telomeres that retained abundant telomeric DNA was somewhat surprising as this phenomenon was not observed in CTC1-deficient mouse cells (39). However as CTC1 binds ssDNA, we reasoned that loss of human CTC1 might allow RPA to accumulate on the G-overhang and/or at replication forks that had stalled within the telomere duplex. Either situation could then lead to ATR activation. Thus, to explore the cause of the DNA damage signaling, we looked for co-localization of RPA with telomere FISH signals on metaphase spreads. Although the RPA staining was weaker than the γH2AX staining, we observed a significant increase in RPA foci at telomeres that retained telomeric DNA (Figure 2D and E). This result was later confirmed by ChIP (see below).
CTC1 deletion causes extensive G-overhang elongation and gradual telomere shortening
To better understand the increase in telomeric RPA, we used non-denaturing in-gel hybridization to examine whether CTC1 disruption causes changes in G-overhang structure and/or an accumulation of ssDNA within the telomere duplex. Genomic DNA from CTC1F/F and CTC1−/− cells was restriction digested, separated briefly in agarose gels and then hybridized with probe to the telomeric G-strand (Figure 3A) or C-strand (Supplementary Figure S3A) under non-denaturing conditions. The DNA was then denatured and re-hybridized with the same probe. To distinguish between ssDNA arising from the G-overhang versus internal regions of ssDNA, control samples were digested with Exo1 prior to restriction digestion and then analyzed in the same gel. Little or no signal was detected by non-denaturing hybridization with either the C-strand probe or with the G-strand probe and Exo1 digested DNA (Figure 3A, Supplementary Figure S3A). Thus, CTC1 deletion does not lead to a significant amount of ssDNA within the telomere duplex region, implying that the build-up of RPA at telomeres is unlikely to be caused by problems with telomere duplex replication. In contrast, when we quantified the amount of G-overhang signal relative to the signal from total telomeric DNA, we found a significant increase in overhang abundance after CTC1 deletion. During the first 7 days after tamoxifen addition, the overhang signal increased ∼3.5 fold and then continued to increase to ∼7-fold by day 14 (Figure 3B). This increase in overhang abundance explains the telomeric RPA accumulation and damage signaling.
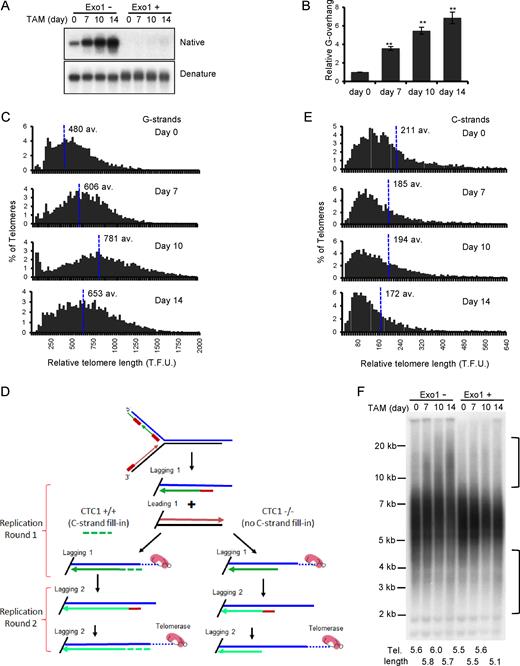
Effect of CTC1 disruption on G-overhang structure and telomere length. CTC1 conditional cells were treated with/without tamoxifen (TAM) for the indicated times. (A and B) Analysis of G-overhang abundance by in-gel hybridization. (A) Gel showing hybridization of TAA(C3TA2)3 probe to genomic DNA under native and denaturing conditions. (B) Quantification of relative G-overhang abundance. Overhang abundance in CTC1−/− cells was normalized to that of CTC1F/F cells (i.e. t = 0). Mean ± S.E.M., n = 3 independent experiments. (C) Analysis of telomere length by FISH. Metaphase spreads were hybridized with (C3TA2)3 telomere G-strand probe and the signal intensity was quantified. Histograms show the distribution of relative telomere lengths expressed as fluorescence intensity (TFU, telomere fluorescence unit), av.; median value, >2000 telomeres were quantified for each sample. A minimum intensity of 100 TFU was set as the cut-off. (D) Cartoon illustrating how loss of C-strand fill-in leads to shortening of lagging strand telomeres following each round of replication. (E) Analysis of telomere length by FISH as in (C) except probe was (G3T2A)3. (F) Southern blot showing length of telomere restriction fragments. Exo1+, DNA was treated with Exo1 prior to restriction digestion. Probe was TAA(C3TA2)3. Brackets indicate telomere that appear to have undergone elongation or shortening. Mean telomere length is indicated below each lane.
The magnitude and progressive nature of the increase in G-overhang abundance after CTC1 deletion was striking because knockdown of STN1 or TEN1 leads to a stable increase of only 1.5–3-fold (6,8,15,19,20). To ensure the large change in overhang abundance was a specific effect of CTC1 removal, we generated CTC1F/F cells that expressed an exogenous FLAG-tagged CTC1 allele (Supplementary Figure S3B) and monitored overhang length in these cells before and after disruption of the endogenous gene locus (Supplementary Figure S3C). Expression of the FLAG-CTC1 completely prevented overhang elongation, indicating that the observed increase in CTC1−/− cells was specific to CTC1 disruption. Thus, it is likely that the lesser change in the STN1 or TEN1 knockdown cells reflects the presence of residual CST because in these cells C-strand fill-in is not completely prevented but is instead delayed until late G2/S or G1 of the next cell cycle, indicating partial CST function (6,19). Consequently, the longer overhangs in the CTC1−/− cells probably reflect more accurately the full length of ssDNA generated during the telomere replication process.
Although CST is thought to limit telomere extension by telomerase (17), depletion of human CTC1, STN1 or TEN1 with shRNA does not necessarily lead to telomere elongation in telomerase positive cells. While elongation is sometimes observed (17), a constant telomere length or slight telomere shortening have also been reported (6,20), suggesting that the effect of depletion depends on the level of knockdown in any specific cell type. To determine how telomere length is affected by complete loss of CTC1, we quantified the telomere FISH signals from metaphase spreads prepared at various times after CTC1 disruption (Figures 2A and 3C). The quantification revealed that CTC1−/− cells undergo both telomere lengthening and shortening with the balance between elongation and attrition changing with time after CTC1 disruption. During the first 7–10 days after tamoxifen addition, some telomeres became much longer and overall median telomere length increased. However, shorter telomeres also accumulated and by day 14 telomere shortening outweighed elongation to cause a decrease in the median length. A similar time-dependent balance between telomere lengthening and shortening was observed when genomic DNA from CTC1−/− cells was analyzed by Southern blotting to visualize the telomeric restriction fragments (TRFs) (Supplementary Figure S3D). During the first 7–12 days of tamoxifen treatment, the TRFs became more heterogeneous with some telomeres showing an increase in length while others showed a decline. At later time points, short TRFs of ≤4 kb gradually became more apparent until by day 21 the average telomere length had declined by ∼0.5 kb. These changes in telomere length were prevented by expression of exogenous FLAG-CTC1 in the CTC1−/− cells (Supplementary Figure S3E).
Given the known roles of CST in telomere duplex replication and C-strand fill-in following telomerase action (6,19,39), it was not hard to rationalize why CTC1 deletion would lead to telomere shortening. Rapid telomere loss could arise from fork stalling during telomere duplex replication and this may partially explain the sudden appearance of the very short telomeres visible in Figure 3C (see day 10). Additionally, gradual telomere attrition is predicted to occur if there is no C-strand fill-in to compensate for incomplete lagging strand synthesis and 5΄ to 3΄ nuclease resection following leading strand synthesis (Figure 3D, Supplementary Figure S3G). This gradual telomere shortening is likely to cause some very short telomeres to become undetectable in the Q-FISH assay (Figure 3C compare days 10 and 14) and hence to be scored as signal free ends (Figure 2A). In contrast to telomere shortening, the apparent telomere growth in the CTC1−/− cells was harder to explain because net elongation of the telomere duplex requires both G-strand synthesis by telomerase and fill-in synthesis of the complementary C-strand by DNA polymerase. Telomerase activity is unaffected by CTC1 loss (Supplementary Figure S3F) and net G-strand elongation takes place in CTC1−/− cells (Figure 3B, see below). But as CST-mediated C-strand fill-in cannot take place, any growth of telomere duplex DNA would require an alternative mechanism to prime replication on the overhang.
The above considerations caused us to question whether the apparent increase in telomere length observed by quantitative FISH (Q-FISH) or Southern hybridization was in fact due to an increase in duplex DNA. As the Q-FISH analysis was performed by hybridizing a probe to the telomeric G-strand, the increase in fluorescence signal at some chromosome ends represents a true increase in the length of the telomeric G-strand. However, the experiment did not assess the length of the telomeric C-strand. Hence, it was possible that the increase in Q-FISH signal and length of the telomeric restriction fragments simply reflected elongation of the G-strand alone. To address this possibility, we repeated the Q-FISH using probe to the telomeric C-strand. The result was striking as we no longer saw telomere elongation at initial time points after CTC1 disruption (Figure 3E). Instead, all samples exhibited a gradual and progressive decrease in telomere length. This finding indicates that loss of CTC1 results in elongation of only the telomeric G-strand.
To confirm the lack of telomere duplex elongation, we treated genomic DNA from the CTC1 conditional cells with Exo1 to remove any extended G-overhangs prior to restriction digestion and visualization of the telomeric restriction fragments by Southern hybridization. The Exo1 treatment caused a dramatic change in the hybridization pattern as all the apparent telomere elongation disappeared but the telomere shortening remained (Figure 3F). Overall these results indicate that loss of CTC1 can result in elongation of the telomeric G-strand in telomerase positive cells, but the elongated G-overhangs cannot then be converted to duplex DNA. As a result, removal of CTC1 leads to telomere shortening because telomere duplex cannot be regenerated after the telomeric C-strand is eroded by nuclease resection and/or incomplete lagging strand replication, or if telomeric dsDNA is lost due to replication fork collapse. The overhang elongation may explain why knockdown of CST subunits has appeared to lead to telomere growth in some situations.
POT1 overexpression partially rescues telomeric DNA damage signaling
While 3΄ overhangs are essential for telomerase action, they pose a danger to the cell due to the ease with which they can elicit DNA damage signals (Figure 2). As a result overhang length and exposure to RPA are tightly regulated. Although this regulation is known to occur through the combined action of CST and POT1/TPP (6,8,19,40), the interplay between these two protein complexes is still poorly understood. We therefore asked whether POT1 can suppress telomeric damage signaling after CTC1 loss.
To answer this question, we over-expressed FLAG-tagged POT1 in CTC1 conditional cells and examined the effect of the elevated POT1 on recruitment of γH2AX and RPA to telomeres. Consistent with previous observations (41), stable expression of FLAG-POT1 suppressed accumulation of endogenous POT1 (Supplementary Figure S4A). Nonetheless, the overall abundance of POT1 in the CTC1F/F cells was elevated 5–10-fold relative to the endogenous level. When FLAG antibody was used to examine POT1 distribution, it was found to localize to telomeres (Figure 4A).
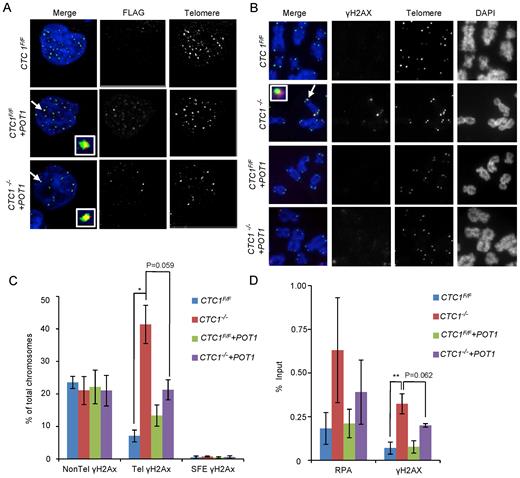
Overexpression of POT1 partially rescues the telomeric DNA damage response. CTC1 conditional cells that did/did not overexpress exogenous POT1-FLAG were monitored for POT1-FLAG, γH2AX and RPA localization to telomeres after growth for 7 days with/without tamoxifen. (A) Localization of POT1-FLAG to telomeres. Interphase cells were analyzed by telomere FISH and FLAG immunostaining. Merge: telomeres (green), FLAG-POT1 (red), nuclei (blue). (B) Localization of γH2AX on metaphase spreads. Telomeres were detected by FISH, γH2AX by immunostaining. Merge: Merge: telomeres (green), γH2AX (red), chromosomes (blue). (C) Quantification of chromosomes with γH2AX foci on chromosome arms (Non-Tel), on one or more telomeres with detectable telomeric DNA (Tel) or at signal free ends (SFE). n = 3 independent experiments, mean ± S.E.M. (D) ChIP analysis showing changes in RPA and γH2AX localization at telomeres after CTC1 loss or POT1 overexpression. Chromatin from the indicated cell lines was precipitated with antibody to RPA or γH2AX. n = >3 experiments for each antibody, mean ± S.E.M.
The effect of POT1 overexpression on DNA damage signaling was first monitored by combined γH2AX immunolocalization and telomere FISH on metaphase spreads of CTC1 conditional cells that did/did not overexpress POT1. The POT1 overexpression caused a reduction in the frequency of telomeric γH2AX staining in the CTC1−/− cells but did not affect the non-telomeric γH2AX (Figure 4B and C). While the reduction in telomeric γH2AX was just below statistical significance, it was highly reproducible (Supplementary Figure S4B). To further assess the effect of POT1 overexpression, we used ChIP to examine both γH2AX and RPA abundance at telomeres (Figure 4D and Supplementary Figure S4C). The ChIP confirmed that CTC1 deletion resulted in both γH2AX and RPA localization to telomeres and showed that this was partially rescued by POT1 overexpression. As with the immunostaining, the rescue by POT1 overexpression was highly reproducible despite the quantification falling slightly below statistical significance (Supplementary Figure S4D). ChIP analysis of TRF2 localization indicated that CTC1 removal or POT1 overexpression caused TRF2 to increase at the telomere (Supplementary Figure S4E), indicating that the telomeric γH2AX staining did not reflect loss of this core shelterin component. The inability of the exogenous POT1 to fully rescue the DNA damage signaling may stem from the lack of TPP1 co-overexpression which would likely limit how much POT1 can accumulate at telomeres.
Given the partial suppression of telomeric DNA damage signaling, we asked whether POT1 overexpression could also rescue the growth of CTC1−/− cells. However, only a very small increase in growth was observed (Supplementary Figure S4F). The lack of rescue may stem from both the residual telomeric DNA damage signaling (Figure 4B–D) and the genome-wide DNA damage in interphase cells previously reported to arise from problems in recovery from replication stress after CST depletion (18,21).
The reduction in telomeric γH2AX and RPA in CTC1−/− cells after POT1 overexpression could reflect the ability of POT1 to outcompete RPA for binding to the long overhangs. However, an alternative possibility is that POT1 suppresses the generation of long overhangs, perhaps by repressing C-strand resection, sequestering the overhang from telomerase extension or somehow promoting C-strand fill-in. To examine these possibilities, we analyzed G-overhang abundance and telomere length in the POT1 overexpressing cells.
G-overhang abundance was monitored by non-denaturing in-gel hybridization as described above, using DNA harvested from CTC1−/− cells after 12 days of tamoxifen treatment (Figure 5A). As before, a large increase in overhang abundance was observed in the CTC1−/− cells. While the POT1 overexpressing cells showed a modest (not statistically significant) decrease in overhang abundance, overall overhang length remained at levels that trigger DNA damage signaling in CTC1−/− cells (see Figure 2B–E, day 7 and Figure 3A, day 7). We therefore conclude that the partial rescue of telomeric DNA damage signaling by POT1 overexpression is most likely because POT1 can compete with RPA to prevent ATR activation (38,40). Our results also imply that the level of ssDNA that can be tolerated before triggering a telomeric DNA damage response is proportional to the cellular level of POT1 or POT1/TPP1.
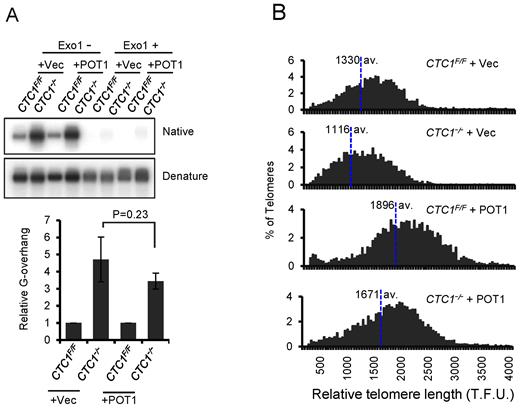
POT1 overexpression does not prevent G-overhang elongation or telomere shortening in CTC1−/− cells. CTC1 conditional cells that did/did not express exogenous FLAG-POT1 were grown with/without tamoxifen for 12 days and assayed for changes in G-overhang abundance and telomere length. (A) Analysis of G-overhang length by in-gel hybridization. Genomic DNA from the indicated cell lines was hybridized with TAA(C3TA2)3 probe first under native conditions then after the DNA had been denatured. Top panel: representative gels. Bottom panel: quantification of G-overhang abundance. Overhang signal from the CTC1−/−cells was normalized to that of CTC1F/F cells. To control for the telomere lengthening caused by POT1 overexpression, overhang signal in the CTC1−/−-POT1 cells was normalized to that of the CTC1F/F-POT1 cells. n = 3 independent experiments, mean ± S.E.M. (B) Quantitative FISH analysis of relative telomere length. Metaphase spreads were hybridized with (C3TA2)3 probe. Histograms show the distribution of relative telomere lengths expressed as fluorescence intensity (TFU, telomere fluorescence unit), >2000 telomeres were quantified for each sample. A minimum intensity of 100 TFU was set as the cut-off.
Telomere length was analyzed by quantitative FISH (Figure 5B) using metaphase spreads and Southern hybridization to telomeric restriction fragments (Supplementary Figure S5). Consistent with previous observations (42) both approaches revealed substantial telomere elongation after POT1 overexpression. This finding indicates that the high levels of POT1 enhanced rather than inhibited telomerase action. Despite having much longer telomeres initially, the POT1 overexpressing cells exhibited substantial telomere shortening after 12 days of tamoxifen treatment. This telomere shortening, in conjunction with the observed G-overhang elongation, indicates that even high levels of POT1 are unable to compensate for loss of CTC1 by mediating C-strand fill-in or preventing C-strand resection.
DISCUSSION
This work provides the first view of how CTC1 deletion, and hence complete loss of the three subunit CST complex, affects the structure and maintenance of human telomeres. We show that loss of CTC1 leads to misregulation of G-overhang processing with accumulation of progressively longer overhangs with increased population doubling. As overhang length increases, POT1 levels become insufficient to prevent RPA from binding the overhang and triggering DNA damage signaling. Although G-overhang length, and overall length of the telomeric G-strand increases, cells lacking CTC1 do not display elongation of the telomere duplex. Rather they exhibit gradual telomere shortening similar to what is observed in cells that lack telomerase activity. However, CTC1−/− cells are competent for telomerase-mediated G-strand synthesis but not the subsequent C-strand fill-in reaction. Thus, the telomere shortening reflects the essential nature of C-strand fill-in for telomere length maintenance. This absolute requirement for C-strand fill-in has important implications for the immortalization of cancer cells through telomerase up-regulation as it indicates that telomere length maintenance, and hence immortalization, will only occur if a cell also has capacity to perform the specialized C-strand fill-in reaction.
C-strand fill-in normally occurs in late S/G2, some hours after most chromosome ends have undergone duplex replication and extension by telomerase (7,9). The requirement for CST in the fill-in process is thought to reflect the need to prime lagging strand synthesis in the absence of the replisome. However, it has been unclear whether conversion of a long G-overhang to duplex DNA can also occur through alternative non CST-mediated priming reactions. These might occur during S-phase of the following cell cycle if the replisome were able to initiate Okazaki fragment synthesis on the ssDNA overhang after finishing replication of the telomere duplex. An alternative post-replicative priming mechanism could utilize PrimPol, a recently discovered DNA primase/polymerase that primes DNA synthesis downstream of UV damage (43). Our results do not rule out some low level of alternative priming reaction. However, they clearly show that if such reactions occur, that they are insufficient to maintain telomere length and CST-mediated priming is essential to prevent progressive telomere shortening and concomitant G-overhang elongation with resultant DNA damage signaling.
It is striking that the loss of a protein complex, which is thought to terminate telomerase action, leads to net telomere shortening rather than telomere elongation. This dichotomy reflects the role of CST in regulating both telomerase and DNA polymerase during G-overhang maturation. Studies with S. pombe indicate that the transition from telomerase-mediated G-strand extension to C-strand fill-in is a multi-step process (44–47) and it is becoming apparent that this is also the case in human cells. Past studies indicate that after POT1/TPP1 recruits telomerase (4), the level of telomeric CST increases in tandem with G-strand extension (17). This most likely occurs because CST binds to the newly synthesized G-strand DNA. Our studies indicate that CST binding prevents RPA accumulation and limits telomerase action. Given that CST can bind POT1/TPP1 (38,48,49), it may be that CST evicts telomerase by disrupting the interaction between telomerase and POT1/TPP1. CST would then enable polymerase α primase (or possibly PrimPol) to engage with the newly synthesized G-strand to initiate C-strand synthesis. The delay in C-strand synthesis is likely to arise because, synonymous to the situation in yeast (45,46,50,51), primase engagement on the G-strand template may depend on cell-cycle regulated posttranslational modification of CST subunits or POT1/TPP1. The above model can explain why loss of CST allows G-strand elongation but not C-strand synthesis or net telomere duplex elongation. It also explains why excess POT1 can partially prevent the elongated overhang from activating a DNA damage response. But as POT1 is not competent to recruit/engage DNA pol α primase, excess POT1 cannot rescue C-strand synthesis.
The role of CST in C-strand fill-in is conserved between mice and humans (6,8,39) and disruption of CTC1 in either organism leads to extensive G-overhang elongation. However, our finding that deletion of human CTC1 mainly results in a robust DNA damage response at telomeres that retain telomeric DNA contrasts with the frequent complete telomere loss and chromosome fusion observed in CTC1−/− mouse cells (39). These differences in outcome are likely to reflect evolutionary changes in the shelterin complex and how it regulates the nucleases responsible for 5΄ end processing. Mouse cells have evolved two POT1 proteins that have only partial overlap in function (8,33). In particular POT1b regulates overhang processing by Apollo, while Pot1a primarily represses ATR signaling. Interestingly, mouse CST interacts with POT1b and this interaction is needed to prevent excess C-strand resection (8). Thus, the complete telomere loss in CTC1−/− mice may result from extensive C-strand resection that occurs when CST is unable to reinforce the protective role of Pot1b. In humans, POT1 may be better able to prevent C-strand resection in the absence of CST such that partial loss of telomere duplex due to replication fork stalling (a likely cause of SFE that lack γH2AX staining) does not then lead to loss of all telomeric DNA.
Going forward, CTC1 conditional human cell lines will provide an important tool for understanding the pleiotrophic nature of Coats plus. To date, analysis of CTC1 patient mutations has only been attempted in human cells that co-express wild-type protein (48), or in a mouse CTC1 knockout cell line (30), making it difficult to ascertain the true in vivo effects of specific mutations. Expression of CTC1 harboring individual patient mutations in CTC1 null human cells will now make it possible to determine which mutations disrupt telomerase regulation and/or C-strand fill-in or the resolution or genome-wide replication stress and hence whether the underlying disease stems from telomeric DNA damage signaling, telomere erosion or general genomic instability. Such information is important to develop effective treatment regimens for patients.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We would like to thank Shiva Senthil Kumar for performing the TRAP assays, Birgit Ehmer for assistance with cell sorting, Anne Forestier for developing protocols, Jason Stewart for helpful comments, Erik Hendrickson for AAV constructs and advice concerning gene targeting, Prasad Jallepalli for advice, Liuh-Yow Chen and Antony Cesare for protocols, Yinhuai Chen and the University of Cincinnati Transgenic Mouse facility for help with design of gene targeting constructs.
FUNDING
National Institutes of Health (NIH) [RO1 GM041803 to C.M.P., F32 F32CA177179, T32 CA117846 to C.H.]. Funding for open access charge: NIH [RO1GM041803].
Conflict of interest statement. None declared.
Present address: Christopher Kasbek, Admera Health, South Plainfield, NJ 07080, USA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments