
Abstract
Recent observations on cancer cell metabolism indicate increased serine synthesis from glucose as a marker of poor prognosis. We have predicted that a fraction of the synthesized serine is routed to a pathway for ATP production. The pathway is composed by reactions from serine synthesis, one-carbon (folate) metabolism and the glycine cleavage system (SOG pathway). Here we show that the SOG pathway is upregulated at the level of gene expression in a subset of human tumors and that its level of expression correlates with gene signatures of cell proliferation and Myc target activation. We have also estimated the SOG pathway metabolic flux in the NCI60 tumor-derived cell lines, using previously reported exchange fluxes and a personalized model of cell metabolism. We find that the estimated rates of reactions in the SOG pathway are highly correlated with the proliferation rates of these cell lines. We also observe that the SOG pathway contributes significantly to the energy requirements of biosynthesis, to the NADPH requirement for fatty acid synthesis and to the synthesis of purines. Finally, when the PC-3 prostate cancer cell line is treated with the antifolate methotrexate, we observe a decrease in the ATP levels, AMP kinase activation and a decrease in ribonucleotides and fatty acids synthesized from [1,2-13C2]-D-glucose as the single tracer. Taken together our results indicate that the SOG pathway activity increases with the rate of cell proliferation and it contributes to the biosynthetic requirements of purines, ATP and NADPH of cancer cells.
Similar content being viewed by others

Main
Cancer cells manifest metabolic alterations that distinguish them from cells in normal tissues,1, 2 which could be exploited for anticancer therapy.3 Both cancer and normal cells rely on glycolysis and oxidative phosphorylation in the mitochondria for energy generation. Oxidative phosphorylation (OxPhos) has a larger energy yield and it is the major pathway in normal cells under normal oxygen tension. However, as first noted by Otto Warburg, cancer cells growing in normal oxygen conditions have an increased rate of glycolysis compared with normal cells (the Warburg effect).4
The fate of increased glucose uptake in cancer cells is a subject of intense research. As noted by Warburg, a significant portion of the consumed glucose is converted to lactate, which is then excreted to the extracellular medium.4 The glycolysis flux can also be diverted from the glycolysis intermediate 3-phosphoglycerate toward serine synthesis, as first demonstrated in lymphomas.5 More recently, it has been reported that the flux toward serine synthesis is upregulated in breast cancer6, 7 and melanoma.8 Studies in melanoma also revealed that a significant portion of the serine produced is converted to glycine.8 Glycine can then enter the glycine cleavage (GC) system, where it is converted to ammonium and methylene tetrahydrofolate (THF), the coenzyme for thymidylate synthesis.5 More recently glycine metabolism has also been shown to be upregulated in non-small cell lung cancers9, 10 and highly proliferating tumor-derived cell lines.11
In parallel to these experimental discoveries, we have recently developed a large-scale model of human cell metabolism that has provided new insights into the advantages of metabolic alterations in cancer cells.12, 13, 14 We have shown that glycolysis has a higher rate of ATP production per mass of pathway enzymes than OxPhos.12, 13 Thus, at high proliferation rates, when the cytosol is overcrowded with ribosomes and metabolic enzymes, glycolysis is more efficient than OxPhos for ATP generation, providing a potential explanation to the evolutionary origin of the Warburg effect. Our model has also predicted that cancer cells can direct their glucose uptake toward a third ATP producing pathway, involving reactions from serine synthesis, one-carbon metabolism and the GC system (SOG pathway, Figure 1).14 The SOG pathway is predicted to be particularly active in tumor cells with decreased pyruvate kinase activity. The relevance of this prediction is underscored by the observation that the pyruvate kinase isoform expressed in most cancer cells (PKM2) can exist in both an active tetrameric form and an inactive dimeric form.15, 16

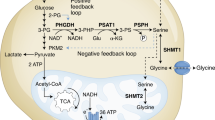
The serine, one-carbon cycle, glycine synthesis (SOG) pathway. Schematic representation of the reactions involved in the SOG pathway, including serine synthesis (green), one-carbon cycle (red) and GC (blue) and the crosstalk with other pathways. The reaction directions are deduced from the inferred fluxes in the NCI60 panel of tumor-derived cell lines (see text for details)
Here we provide additional evidence for the activity of the SOG pathway in a subset of human cancers. To this end we interrogate gene expression profiles for different cancer types and metabolic fluxes for the NCI60 panel of cancer cell lines. We also investigate the SOG pathway inhibition by the antifolate methotrexate, focusing on signatures of energy stress and changes in metabolic fluxes using a targeted tracer fate association study (TTFAS).17, 18
Results
The predicted SOG pathway
The predicted pathway14 is depicted in Figure 1. It includes the synthesis of serine from 3-phosphoglycerate with the concomitant conversion of glutamate into α-ketoglutarate (Figure 1, serine synthesis). Serine is then converted to glycine by the cytosolic serine hydroxymethyltransferase (SHMT1) with concomitant conversion of THF into 5,10-methylene-THF (CH2-THF). Alternatively, serine can be transported to the mitochondria where it can be converted to glycine by the mitochondrial serine hydroxymethyltransferase (SHMT2). Glycine can be excreted or enter the cleavage system, where it is converted to ammonium with the concomitant conversion of another THF molecule into CH2-THF (Figure 1, GC). CH2-THF can be transformed back to THF either in the cytosol or in the mitochondria with the concomitant generation of two ATPs (Figure 1, one-carbon cycle). In the cytosol this is carried out by the tri-functional enzyme MTHFD1, with CH2-THF dehydrogenase (NADP dependent), 5,10-methenyl-THF (CH+-THF) cyclohydrolase and 10-formyl-THF (CHO-THF) synthetase activities.19 In the mitochondria, these activities are partitioned between MTHFD2, with CH2-THF dehydrogenase (NAD dependent) and CH+-THF cyclohydrolase activities;20 MTHFD2L, with CH2-THF dehydrogenase (NADP dependent);21 and MTHFD1L, with CHO-THF synthetase activity.22
Excluding cofactors (NAD+, glutamate, NADP+ and ADP) and products (NADH, α-ketoglutarate, NADPH, CO2, NH4+, formate and ATP), this pathway is stoichiometrically balanced and produces two ATPs per molecule of 3-phosphoglycerate, four ATPs per molecule of glucose. The ATP producing step is catalyzed by the reverse CHO-THF synthetase activity of MTHFD1 or MTHFD1L. Kinetic modeling of the one-carbon cycle demonstrates that operation of this cycle in the ATP direction is thermodynamically feasible.14 We also note that imported serine can also be utilized to run the one-carbon cycle, with a yield of two ATPs or one ATP per molecule of serine, depending on whether glycine is or is not cleaved, respectively. Similarly, glycine imported from the environment can be utilized to run the one-carbon cycle as well, with a yield of one ATP per molecule of glycine.
Intermediate metabolites of the SOG pathway crosstalk with other pathways as well (Figure 1, cyan-colored lines). Serine and glycine are utilized for protein synthesis and glycine is required for purine synthesis as well. CH2-THF is required for thymidylate (dTMP) and methionine (Met) synthesis, and CHO-THF is required for the de novo synthesis of purines (IMP). Finally, the NADP+-dependent CH2-THF dehydrogenase activities of MTHFD1 and MTHFD2L may contribute to the NADPH production in the cytosol and the mitochondria, respectively.23
SOG pathway gene expression in human cancers
To investigate whether the SOG pathway is expressed in human cancers, we performed gene signature analyses across several cancers. The genes associated with this pathway are PHGDH, PSAT1 and PSPH encoding their respective enzymes involved in serine synthesis, SHMT1, SHMT2, MTHFD1, MTHFD2, MTHFD2L and MTHFD1L encoding their respective enzymes involved in the one-carbon cycle, and GCSH, DLD, GLDC and AMT encoding the H, L, P and T proteins of the GC system, respectively (Figure 1). These genes were compiled into a gene signature that we then used to interrogate their expression across cancers. To quantify the prevalence of this signature in human cancers, we performed a gene set enrichment analysis (GSEA).24 For each sample analyzed, GSEA reports a score and a statistical significance (p) for upregulation and downregulation of the gene signature.
As shown in Figure 2, the gene signature scores capture the concordant overexpression or under-expression of the pathway genes in the samples analyzed. It is evident that there is a subset of breast- (Figure 2a) and prostate cancers (Figure 2b) where most genes in the SOG pathway are highly expressed, resulting in a significant expression of the SOG gene signature. Extending this analysis to other cancer types, we observe that about 28% of lung-, 19% of breast-, 9% of prostate-, 30% of colorectal-, 23% of brain- and 21% of ovarian cancers manifest a significant upregulation (P<0.05) of the SOG pathway gene signature (data not shown).

Expression of genes encoding for enzymes in the SOG pathway in human cancers, differentiated cells and embryonic stem cells. Expression of genes encoding for enzymes in the SOG pathway (rows) across (a) breast and (b) prostate cancer patients (columns), based on data from50 and,51 respectively. The top rows display the SOG pathway, Myc target activation and proliferation gene signature upregulation scores as quantified by GSEA, together with the Myc gene expression. Blue color denotes under-expression and red color represents overexpression. The upper bar indicates the subset of tumors, where the SOG pathway signature is significantly downregulated (P<0.05, down), intermediate or significantly upregulated (P<0.05, up). (c) Expression of genes encoding for enzymes in the SOG pathway in human embryonic and differentiated normal cells, based on data reported in.30 (d) Expression of genes encoding for enzymes in the SOG pathway during differentiation of mouse R1 embryonic stem cell, based on data reported in.32 The samples are labeled by the day from initiation and by the condition used: embryonic body (EB) formation in non-adherent plastic dishes, gelatin coated plates (GEL) and matrigel coated plates (MAT). The white squares represent missing expression data
Next, we investigated the association of the SOG pathway gene signature with markers of poor prognosis. In breast cancers, the SOG pathway signature is significantly correlated with proliferation (Pearson Correlation Coefficient, PCC=0.62, statistical significance P=1 × 10−6), Myc gene expression (PCC=0.64, P=1 × 10−6), a gene signature of Myc targets expression (PCC=0.58, P=1 × 10–6) (Figure 2a) and estrogen receptor-negative (ER−) status (PCC=0.46, P=1 × 10–6), known markers of poor prognosis in breast cancer.25, 26 In prostate cancers, the pathway signature is also positively correlated with markers of malignancy, including proliferation (PCC=0.38, P=1 × 10–6), marginally with Myc gene expression (PCC=0.16, P=1.3 × 10–3), Myc targets expression (PCC=0.44, P=3 × 10–6) (Figure 2b), TMPRSS2-ERG fusion (PCC=0.34, P=1 × 10–5) and Gleason score (PCC=0.18, P=1 × 10–3). Finally, in a pooled analysis of 3947 samples from multiple cancer types, we corroborated the association of the pathway signature with increased proliferation (PCC=0.50, P=2 × 10–6) and the gene signature of Myc targets expression (PCC=0.49, P=2 × 10–6).
Taken together these data indicate that the expression of the SOG pathway genes is correlated with gene signatures of cell proliferation and Myc activation in human cancers. This observation is in agreement with previous work analyzing in vitro data for tumor-derived cell lines11 and a mouse model of Myc driven tumor formation in the liver.14, 27 Furthermore, we recall that Myc amplification and/or overexpression are common events in human cancers,28 which would explain the correlation between Myc gene expression, Myc targets gene expression (including SOG pathway genes) and proliferation.
SOG pathway gene expression in embryonic stem cells
The metabolic alterations characteristic of cancer cells are often observed during embryonic development as well.29 Thus, we hypothesized that the SOG pathway may be active in embryonic stem cells. To test this hypothesis, we analyzed reported gene expression profiles for embryonic and differentiated cells.30 We observe that most genes in the SOG pathway are highly expressed in embryonic stem cells and embryonic bodies relative to differentiated cells (Figure 2c). We also observe some differences regarding the genes MTHFD1 and MTHFD2 encoding for the cytosolic and mitochondrial one-carbon metabolism enzymes, respectively. The expression profiles indicate that the mitochondrial isoenzyme is overexpressed in embryonic cells, whereas the cytosolic isoform is overexpressed in leukocytes and stromal cells (Figure 2c). Finally, in the case of PSPH there is not a significant difference between embryonic and differentiated cells (Figure 2c), suggesting that this enzyme is not responsible for the regulation of the activity of the SOG pathway at the gene expression level. We also observe that upregulation of the SOG pathway genes coincides with upregulation of gene expression signatures for Myc targets activation and increased proliferation. The former is consistent with the fact that many of the SOG pathway genes are Myc targets (PHGDH, PSPH, SHMT1, MTHFD1, MTHFD2 and GCSH14, 31) and the latter with the fact that embryonic stem cells manifest high proliferation rates. These observations are corroborated by the analysis of a second data set reporting gene expression profiles during differentiation of a mouse embryonic stem cell.32 Following the time course of differentiation, we observe that the SOG pathway genes are upregulated in the initial steps of embryonic development (Figure 2d, Day 0–3). In contrast, the SOG pathway genes are downregulated following differentiation (Figure 2d, Day 5 and 7). As in the previous case, we confirm that the upregulation of SOG pathway genes coincides with the upregulation of signatures for Myc targets activation and increased proliferation.
We note that the gene encoding for dihydroproline dehydrogenase (DLD in humans and Dld in mouse) manifests an opposite pattern compared with the other genes, that is, it is downregulated in embryonic cells and upregulated in differentiated cells. This gene encodes for the L protein of the GC system. The L protein is also a component of the pyruvate dehydrogenase33 and the α-ketoglutarate dehydrogenease34 complexes, two key enzymes of the TCA cycle. The gene expression data indicate that the expression of dihydroproline dehydrogenase correlates more with its function as a component of the TCA cycle than the GC system. We also note that DLD is one of the exceptions of the SOG pathway genes that are not annotated as Myc targets.
Although the Myc activation signature is upregulated in embryonic stem cells and downregulated in differentiated cells, the gene expression of Myc itself is not associated with the differentiation status during embryonic development (Figures 2c and d). The absence of a consistent correlation between the Myc gene expression and the expression of Myc targets suggests a post-translational mode of Myc regulation. Post-translational regulation of Myc has been observed in differentiated muscle cells, through the proteolytic cleavage of the Myc protein by calcium-dependent calpains in the cytosol.35 Thus, in embryonic stem cells Myc is likely activated at the post-translational level, resulting in an increased expression of Myc target genes (including SOG pathway genes) and increased proliferation.
SOG pathway inferred metabolic fluxes in tumor-derived cell lines
To investigate the correlation between the SOG pathway activity and proliferation at the level of metabolic fluxes, we next investigated previously reported exchange fluxes for the NCI60 panel of tumor-derived cell lines.11 We developed ‘personalized’ metabolic models to infer the internal metabolic fluxes that are consistent with the reported exchange fluxes (Supplementary Methods). The personalization to each cell line included the doubling time, the cell volume and the DNA content. The model was validated at two different levels. At the total protein content level, the model predicted that protein content for each cell line is in good agreement with the measured value (Supplementary Methods, Supplementary Figure S1). At the level of each reaction, we compared the model predicted flux with the expression of proteins that are part of the enzyme complex catalyzing the reaction. The protein expression values were obtained from a recent proteomic analysis of the NCI60 cell lines.36 A total of 1578 proteins were linked to reactions in the metabolic model based on the BiGG database annotations37 (Supplementary Table 1). In 41% of the cases there is an agreement between the protein being expressed and the associated reaction found active in one or more cell lines. This group is enriched for metabolic pathways associated with the central metabolism, including glycolysis, OxPhos, serine, glycine and folate metabolism (Supplementary Table 2, Type 1). In 7% of the cases there is also an agreement between the protein not being expressed and the associated reaction not found active in any cell line (Supplementary Table 2, Type 4). In contrast, in 51% of the cases the protein is detected in one or more cell lines but the model predicts a zero flux for the associated reaction in all cell lines (Supplementary Table 2, Type 3). This group is enriched for pathways associated with the metabolism of fatty acids and some amino acids, and with reactions taking place in the lysosome and peroxisomes (Supplementary Table 2, Type 3). These are areas were the model will require future improvements. Finally, in just 2% of the cases the protein is not detected in any cell line but the model predicts a non-zero flux for the associated reaction in at least one cell line (Supplementary Table 2, Type 2).
Among the instances where the protein was detected and the reaction was found active the protein is detected in one or more cell lines but the model predicts a zero flux for the associated reaction in all cell lines (Supplementary Table 2, Type 1), in 9% of the cases there is a significant correlation (P<0.05) between the protein expression and the reaction flux for each cancer cell line. This is a reasonable value given that each protein can be part of an enzymatic complex whose final expression is a non-trivial function of the expression of its components. For example, the expression of only one (out of 11, 9%) and only two (out of 30, 7%) subunits of ATP synthase and NADH dehydrogenase, respectively, correlates with their predicted reaction rates (Supplementary Table 1). Furthermore, reaction rates may be controlled by post-translational regulation, metabolite concentrations and the rate of the associated metabolic pathway.38 For example, metabolic control analysis indicates that, in highly proliferating cells, serine synthesis from 3-phosphoglycerate is controlled by the serine demand rather than the specific expression levels of each enzyme on the pathway.39
Using these personalized metabolic models, we have estimated the fluxes of reactions in the SOG pathway. To avoid biases due to cell size variations across the NCI60 panel, we have normalized fluxes by cell volume. The estimated fluxes of reactions in the SOG pathway are highly correlated with proliferation rates (Figure 3). Specifically, the flux of serine synthesis from 3-phosphoglycerate exhibits a positive correlation with the proliferation rate (PCC=0.24, P=0.034, Figure 3a). The serine synthesized from glucose together with that imported from the culture media (Figure 3b) are in part converted to glycine via a SHMT activity, in a magnitude that increases with increasing the proliferation rate (PCC=0.77, P<10–6, Figure 3c). This flux is distributed between both the cytosolic (SHMT1, PCC=0.77, P<10–6, Figure 3d) and the mitochondrial (SHMT2, PCC=0.64, P<10–6, Figure 3d) SHMTs. In slowly proliferating cells, the produced glycine exceeds the biosynthetic needs of the cell and it is excreted (Figure 3e), as previously reported.11 In several highly proliferating cell lines, the glycine produced from serine together with glycine imported from the media are in part catabolized through the GC system, overall resulting in a positive correlation between the GC flux and the proliferation rate (PCC=0.41, P=0.00056, Figure 3f). The THF consumed in the conversion of serine to glycine and in the GC is produced through the one-carbon cycle, with a concomitant generation of ATP, with a net median MTHFD (MTHFD1+MTHFD1L) flux that increases with the increasing proliferation rate (PCC=0.70, P<10–6, Figure 3g). However, we observe noticeable differences between the cytosolic and mitochondrial cycles. In the cytosol, MTHFD1 is predicted to work in the direction of THF and ATP production (PCC=0.74, P<10–6, Figure 3h), whereas MTHFD1L is predicted to work in the opposite direction in the mitochondria (PCC=0.73, P<10–6, Figure 3h). We also note that the MTHFD2 cyclohydrolase activity contributes to the production of CHO-THF in the mitochondria, at a magnitude that increases with the increasing proliferation rate (PCC=0.64, P<10–6, Figure 3i). In the case of GC (Figure 3f) and the MTHFD net flux (Figure 3g) the predicted 90% confidence intervals expand a wide range. Therefore, we cannot exclude the possibility that for some atypical choice of kinetic parameters glycine is not catabolized or there is no ATP production from the one-carbon cycle.

Metabolic fluxes as a function of the proliferation rate. (a–i) Estimated metabolic fluxes of reactions in the SOG pathway are shown as a function of the proliferation rates of cell lines. Each point/error bar represents a cell line in the NCI60 panel. The point represents the median over the range of model kinetic parameters explored, and the error bars represent the 90% confidence intervals. The dashed lines are linear fits. Panels (b and e) report exchange fluxes as measured in.11 These fluxes are fixed in the personalized models and, therefore, they do not display error bars due to variations in the model kinetic parameters
Crosstalk with other metabolic pathways in tumor-derived cell lines
Protein synthesis
Some SOG pathway intermediate metabolites are precursors for biosynthetic processes. Serine, glycine and methionine are required for protein synthesis. The sum of imported serine and glycine are of the same amount as is required for protein synthesis (data not shown), indicating that additional serine synthesized from glucose fulfills a different requirement. The serine synthesis rate from glucose is actually higher than the serine import rate (Figure 4a). This extra serine is converted to glycine (as discussed above, Figure 3c), fueling the SOG pathway.

Crosstalk with other pathways. (a–i) Estimated rates of selected reactions that are part or crosstalk with the SOG pathway as a function of the proliferation rates of cell lines. Each point/error bar represents a cell line in the NCI60 panel. The point represents the median over the range of kinetic parameters explored and the error bars represent the 90% confidence intervals
Glutamate→α-ketoglurate
In an intermediate step of serine synthesis from 3-phosphoglycerate, the cytosolic transaminase PSAT converts glutamate to α-ketoglutarate, at a rate that is about half of the total net conversion of glutamate to α-ketoglutarate (Figure 4b). This result is in agreement with previous labeled-carbon trace experiments showing a conversion of glutamate to α-ketoglutarate linked to serine synthesis.6
ATP balance
The rate of ATP production by the cytosolic MTHFD1 is comparable to that inferred for pyruvate kinase (Figure 4c), indicating that the SOG pathway has a significant contribution to ATP production in the cytosol. In contrast, the mitochondrial MTHFD1L consumes ATP. This consumption is balanced by the ATP production of ATP synthase (Figure 4d).
NADPH balance
In the mitochondria, the NADP+-dependent CH2-THF dehydrogenase activity of MTHFD2L contributes to NADPH production in an amount comparable to glutamate dehydrogenase (GLUD, Figure 4e). The overall NADPH produced from the combined activities of MTHFD2L and GLUD is balanced by the activity of nicotinamide nucleotide transhydrogenase (NNT) in the direction of NADPH oxidation (Figure 4f), with the concomitant translocation of protons from the mitochondria to the cytosol (H+, Figure 1). These protons are translocated back from the cytosol to the mitochondria by the ATP synthase activity, with concomitant ATP production (Figures 1 and 4d). In the cytosol, the NADP+-dependent CH2-THF dehydrogenase activity of MTHFD1 contributes to NADPH production in an amount comparable to that of glucose-6-phosphate dehydrogenase (G6PD, Figure 4g) and to the NADPH requirements of fatty acid synthase (FAS) (Figure 4h).
Purines, methionine and thymidylate
In the de novo pathway of purine synthesis both ATIC and GART convert CHO-THF to THF. The added rate of ATIC+GART reaches maximum values close to 0.02 pmol/pl/h (Figure 4i). These values are comparable to the NADPH production rates that also reach maximum values around 0.02 pmol/pl/h (Figure 4g). The ATIC+GART maximum rates are, however, 10-fold lower than the maximal ATP production rates from MTHFD1, which reach average values around 0.2 pmol/pl/h (Figure 4c). Although there is a non-zero rate of thymidylate synthase (TYMS) and of methionine synthase (MS), their values are relatively smaller than the reaction rates described above (Figure 4i).
Treatment with methotrexate causes energy stress and inhibits purine and fatty acid synthesis
The SOG pathway contains reactions metabolizing folate derivatives and, therefore, may be subject to inhibition by antifolates such as methotrexate (Figure 1). A reduction in the concentration of folate derivatives below the half-saturation constants of enzymes in the one-carbon cycle would result in a decrease of their rates. Methotrexate inhibits the activity of dihydrofolate reductase (DHFR, Figure 1), resulting in a decrease of the THF pool. On the basis of these observations, we hypothesized that treatment with methotrexate should inhibit the predicted activities of the SOG pathway. To test this hypothesis, we investigated the response of PC-3 cells to treatment with methotrexate.
Treatment with methotrexate inhibits the proliferation of PC-3 cells in a dose-dependent manner (Figure 5a). At 100 nM, a concentration above the IC50 for this cell line (30 nM), we observe an initial phase of growth inhibition within the first 24 h, followed by a cytotoxicity characterized by a significant reduction of cell numbers (Figure 5a). We hypothesized that the inhibition of the SOG pathway ATP production may be a mechanism of growth inhibition in the first 24 h following treatment. To test this hypothesis, we measured changes in the levels of the adenine nucleotides AMP, ADP and ATP. Four hours after treatment, there is a significant decrease in the ATP levels relative to those at 2 h after treatment (Figure 5b, 4 h, P=0.0061). In contrast, at this time point, there was a lesser decrease in the ADP levels (Figure 5c, 4 h, P=0.031) and no significant change in the AMP levels (Figure 5d, 4 h, P=0.29). The drop in ATP levels at 4 h, with no significant change in the AMP levels, results in a higher AMP/ATP ratio (Figure 5f). An increase in the AMP/ATP ratio is expected to activate the AMP kinase (AMPK), due to both increased AMPK phosphorylation and the allosteric activation by AMP itself.40 To investigate the activation of AMPK, we measured the levels of AMPK phosphorylation as well as phosphorylation of acetyl-CoA carboxylase (ACC), a known AMPK target.40 The levels of phosphorylated AMPK (pAMPK) increase by threefold 2 h after treatment (Figures 5g and h). Similarly, with some time lag, we observe a threefold increase of phosphorylated ACC (pACC) (Figures 5g and h). Taken together, the drop in ATP levels and the increase of pAMPK and pACC indicate energy stress as part of the initial response (0–4 h) to methotrexate treatment.

The effect of methotrexate treatment on metabolic parameters. (a) PC-3 cell count as a function of time in cell cultures untreated and treated with 10 and 100 nM of methotrexate. (b–f) Changes in (b) ATP, (c) ADP and (d) AMP levels and in (e) ADP/ATP and (f) AMP/ATP ratios following treatment with 100 nM methotrexate. (g) Western blots of AMPK, pAMPK, ACC and pACC following treatment with 100 nM methotrexate. (h) Quantification of the pAMPK/AMPK and pACC/ACC ratio relative to control (time zero data). (i) EZTopolome (heatmap) summarizing the changes in labeled metabolites measured in triplicate, in cell cultures untreated (control) and treated with 100 nM methotrexate (MTX). Red/blue indicates expression above/below the mean across samples. The top row indicates the color code for expression level changes
Subsequently, at the 8-h time point, in addition to the ATP levels (Figure 5b, 8 h, P<10–6), the ADP levels have decreased significantly (Figure 5c, 8 h, P=0.0087), whereas the AMP levels have not changed significantly yet (Figure 5d, 8 h, P=0.37). Finally, at 24 h, the levels of all nucleotides have decreased significantly (Figures 5b–d, 8 h; ATP, P=0.00096; ADP, P=0.0011; AMP, P=0.031). This observation indicates the inhibition of adenine nucleotide synthesis as part of the response to methotrexate at intermediate times (8–24 h).
Next, we performed targeted [1,2-13C2]-D-glucose fate association experiments to investigate changes in metabolic fluxes. PC-3 cells were grown in a medium containing this precise glucose tracer,41 untreated or treated with methotrexate, and the concentration of different labeled metabolites were measured 24 h after treatment (Figure 5i). There was no substantial difference in the glucose consumption (treated/untreated=0.93) and the medium 13C-lactate fraction (treated/untreated =0.96), indicating that there are no major changes of the glycolysis rate. There is no substantial difference in the released 13CO2 fraction (treated/untreated=0.99) and only a marginal decrease in the media 13C-glutamate fraction in the culture medium (treated/untreated=0.80, P=0.050), indicating that there are no significant changes in the rate of OxPhos. We observe a 1.5-fold decrease in the intracellular 13C-RNA-Ribose fraction (P=0.050), which is consistent with the inhibition of purine synthesis by methotrexate.42, 43 We also observe a 2.1-fold decrease in the intracellular 13C-palmitate fraction (P=0.050). The reduction in palmitate synthesis could be explained by an inhibition of the NADPH production from CH2-THF dehydrogenase activity of MTHFD1. The reduction in palmitate synthesis is also consistent with the reported increase in ACC phosphorylation following methotrexate treatment. ACC is a key enzyme in fatty acid synthesis and phosphorylation inhibits its enzymatic activity.
Coming back to the initial ATP depletion, we further note that the ATP levels drop about 10 μmol/g protein from 2 to 4 hours, indicating an unbalance between production and consumption of ATP of about 5 μmol/g protein/h. The targeted trace experiments indicate that there are no significant changes in the rate of glycolysis and OxPhos in treated versus untreated PC-3 cells, ruling out the inhibition of these two pathways as a cause of the ATP depletion. It is also unlikely that the initial drop in ATP levels occurs solely due to a decrease in purine synthesis. The rate of purine synthesis in untreated PC-3 cell lines, as estimated from the rate of GART or ATIC, is 1.4 μmol/g protein/h, that is, fivefold lower. Furthermore, the levels of the other purines ADP and AMP do not manifest a drop as pronounced as that for ATP in the first 4 h. On the other hand, the estimated ATP production rate from MTHFD1 in untreated PC-3 cells is 34 μmol/g protein/h, albeit with an error bar of the same magnitude. Taken together these data indicate that an inhibition of the ATP production by MTHFD1, due to relative changes in the folate pool, is the most likely cause of the initial drop in the ATP levels. In contrast, the subsequent decrease in ATP levels has a slower kinetics, decreasing about 5 μmol/g protein from 4 to 24 h, that is, at a rate of 0.25 μmol/g protein/h. This decay is more likely to be caused by the inhibition of purine synthesis, as both the ATP and ADP concentrations manifest a similar decrease within this time frame.
Discussion
The analysis of gene expression data across human cancers indicates that the expression of genes coding for enzymes in the SOG pathway is correlated with gene signatures of cell proliferation and Myc target activation, as previously shown in vitro.11 Several genes in the SOG pathway are Myc targets and their expression increases upon Myc activation in an inducible Myc model of murine liver cancer.14 The association between SOG pathway activity and cell proliferation is further supported at the level of metabolic fluxes of cancer cell lines grown in vitro. The estimated fluxes of serine synthesis from glucose, serine import, serine conversion to glycine and GC are all correlated with the proliferation rate of the cell lines in the NCI60 panel. We have previously shown that sensitivity to the antifolate methotrexate is associated with the expression of the SOG pathway mitochondrial enzymes, in vitro and in the treatment of acute lymphocytic leukemia (ALL).44 This observation is concordant with the correlation between proliferation and the activity of the SOG pathway and the known specificity of methotrexate against highly proliferating cells.
The analysis of the inferred fluxes for the NCI60 panel of tumor-derived cell lines indicates that the bulk of the SOG pathway flux is directed to the ATP, NADPH and purine requirements of biosynthesis. The contribution of folate metabolism to purine synthesis has been studied extensively.43, 45 The contribution to ATP production supports our previous theoretical prediction implicating the SOG pathway in energy generation.14 Here, we also uncover the contribution of this pathway to the NADPH requirements of cell proliferation, for example, for fatty acid synthesis. We also observe that these three major functions of the SOG pathway are inhibited by treatment with the antifolate drug methotrexate. The inhibition of ATP production is demonstrated by a fast initial decay in the ATP levels. The inhibition of NADPH production is deduced from a reduction in fatty acid synthesis from glucose. The inhibition of purine synthesis is demonstrated by a reduction of adenine nucleotide levels and of ribonucleotide synthesis from glucose following methotrexate treatment.
The estimated SOG pathway flux in tumor-derived cell lines differs from the standard view of the folate cycle:45 (i) The exchange of carbon units between the cytosol and the mitochondria takes place via the transport of serine, glycine and formate through the mitochondrial membrane. (ii) The mitochondrial MTHFD1L converts CHO-THF into THF with concomitant formate production. (iii) The cytosolic MTHFD1 converts THF to CHO-THF consuming the formate produced in the mitochondria. To our knowledge, there is no definitive evidence supporting this view in human cells. The best available data are L-[3-13C]-serine and [13C]-formate tracer experiments using the breast cancer cell line MCF-7.46 When MCF-7 cells were grown on (L-[3-13C]-serine), ([13C]-formate), (L-[3-12C]-serine, [13C]-formate) and (L-[3-13C]-serine, [12C]-formate), there was about 95%, 80%, 70% and 32% 13C-labeling of purines, respectively. These data have been interpreted as formate being the major donor of one-carbon units for purine synthesis in the cytosol and formate being synthesized from serine in the mitochondria. However, that data are also consistent with the folate cycle reported here (Figure 1), where both serine and formate are transported to the mitochondria and donate one carbon to CHO-THF, which is then transported to the cytosol where it is utilized for purine synthesis. We note that the activity of the mitochondrial folate transporter (MFT) is a key component of the latter hypothesis. It has been shown that reduced folates (e.g., THF and CHO-THF) can be transported across the mitochondrial membrane and that this activity is in part mediated by the MFT.47, 48 However, this observation has not been taken into account in the standard view of the cytosol-mitochondria folate cycle. Further quantification of the labeled fractions of folate derivatives in the cytosol and the mitochondria is therefore required to discriminate between the standard view of the folate cycle and the alternative view proposed here (Figure 1).
Materials and Methods
Microarray data and analyses
The breast cancer data set included 295 samples previously reported by van’t Veer et al.49 and van de Vijver et al.50 The prostate cancer data set was from 281 men with localized prostate cancers previously reported by Sboner et al.51 with gene expression measured using the Human 6 k Transcriptionally Informative Gene Panel for DASL.51 Pooled analysis of cancer data sets was carried out using the two data sets listed above and the following data sets from the Gene Expression Omnibus (GEO) repository (Accessed December 2011); colon cancer GSE17536, GSE14333 and GSE20916; brain cancer GSE4271, GSE4290 and the NCI Rembrandt database; lung cancer GSE31210, GSE11969 and GSE19188; and ovarian cancer GSE9899 and GSE6008. Data collection and normalization are described in Supplementary Methods. The SOG pathway signature is reported in the Results section. The remaining gene signatures were obtained from: proliferation,52 embryonic stem cell53 and Myc activation.31 The signature scores and statistical significance were calculated as previously described.24
Personalized metabolic models
Personalized metabolic models were developed for each cell line in the NCI60 panel, as described in Supplementary Methods. The personalization included previously reported exchange fluxes,11 DNA content estimates based on previously reported karyotypes and the cell volume. The cell volume of each NCI60-panel cell line was estimated using cell size measurements obtained from Cellometer Auto T4 (Nexcelom Bioscience LLC, Lawrence, MA, USA).54 The image cytometer utilizes a bright-field (BR) light microscopy optical setup for image cytometric analysis. Each cell line was pipetted into the disposable counting chamber and bright-field images were captured for image analysis, where the system measured the pixel area of the cells and converted the results to corresponding cell diameter. The image cytometer operation and functionalities have been described previously.55, 56
Adenine nucleotide measurements
Methotrexate treatment: 1.5 × 106 PC-3 cells were seeded in triplicate for each time point in 100 mm dishes overnight in RPMI medium supplemented with 10% dialyzed FBS and placed in a 37 °C incubator with 5% CO2. Twenty-four hours after seeding, culture media were removed and replaced with media containing 100 nM methotrexate or methotrexate-free media and placed back into a 37 °C incubator. For collection at each time point, the culture media was remove and the plate was washed three times with cold Ca++, Mg++ and phenol red free HBSS (Gibco, Grand Island, NY, USA). With the plate on ice, 0.5 ml of 0.6 N perchloric acid was added and the plate was quickly scraped and collected into a microcentrifuge tube. The plate was scraped again with another 0.5 ml of 0.6 N perchloric acid and both portions were combined. Each sample was homogenized using a Polytron PT 1300 homogenizer at 7000r.p.m. for 30 s on ice. Samples were centrifuged at 13 000 × g at 4 °C for 10 m to pellet cell debris. The supernatant was transferred to a clean microcentrifuge tube and stored at −80 °C until analysis. Cell pellets were lysed in 1.0 ml 0.2N NaOH for protein assay using the Bradford method.
The quantification of intracellular adenine nucleotides was performed using a fluorometric method as described.57 The cellular extracts and the standards (ATP, ADP and AMP prepared in 0.6N perchloric acid) were neutralized with 0.8 ml ice-cold Freon/trioctylamine (4 : 1, by vol). The neutralized nucleotide extract was vortexed for 30 s, then centrifuged at 13 000 × g for 5 m at 4 °C. The upper aqueous layer was collected for derivatization of nucleotides into fluorescent 1,N6-etheno derivatives. A 50 μl aliquot of nucleotide extract was mixed with 150 μl freshly prepared mix of chloroacetaldehyde (50%), to acetate buffer (1M, pH4.5) (11.2 : 138.8, v/v) in a 1.5 ml microcentrifuge tube, mixed and then heated at 60 °C for 1 h. After the reaction, the tubes were placed on ice to stop the reaction. The samples were diluted five times and 50 μl of the derivatized sample was used for adenine nucleotide analysis and the results were expressed as μmol/g protein.
Quantification of adenine nucleotide was performed using a high-performance liquid chromatographic system (Hitachi, Tokyo, Japan) equipped with an L-7200 autosampler tray maintained at about 8 °C, L-7100 pump and L-7480 fluorescence detector with excitation and emission wavelengths set at 280 nm and 410 nm, respectively. The samples and standards were analyzed using a Waters Sunfire ODS column (5 μm, 250 × 4.6 mm, Milford, MA, USA) at 30 °C, and the mobile phase A: 30 mM KH2PO4+0.8 mM TBAP (tetrabutylammonium phosphate), pH 5.45; and B: acetonitrile/30 mM KH2PO4 (1 : 1, by vol.)+0.8 mM TBAP, pH 7.0. The analytes were separated using gradient elution at 1 ml/min.57 The identity of etheno-adenine nucleotides was determined by comparing retention times to known nucleotide standards. The retention times of adenine nucleotide derivatives AMP, ADP and ATP were 11 min, 19 min and 21 min, respectively. The concentrations of adenine nucleotides in the cells were calculated from the standard curve by linear regression analysis.
AMPK activation
Twenty-four hours before methotrexate treatment, 7.5 × 105 PC-3 cells were seeded in six-well plates in RPMI medium supplemented with 10% dialyzed FBS and incubated overnight at 37 °C in an incubator with 5% CO2. At the time of treatment, culture media was remove and replaced with fresh media containing 100 nM methotrexate or methotrexate-free media and placed into a 37 °C incubator with 5% CO2. At the appropriate time points, culture media was remove; cells were rinsed with PBS (Gibco) and scraped into microcentrifuge tubes. After brief centrifugation, cell pellets were lysed in RIPA buffer containing a commercial protease inhibitor mix (Roche, Nutley, NJ, USA) and phosphatase inhibitor (50 mM sodium fluoride 10 mM sodium orthovanadate). After protein quantification by the Bradford protein assay (Bio-Rad Laboratories, Hercules, CA, USA), proteins were resolved by 10% SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad Laboratories). After blocking the membrane with 5% nonfat dry milk prepared in tris-buffered saline+0.1% Tween-20, the membrane was incubated with the desired primary antibody according to the manufacturer’s directions at 4 °C overnight. The membrane was washed in tris-buffered saline+0.1% Tween-20 and incubated for 2 h at room temperature with the appropriate peroxidase-conjugated secondary antibody. Bands were visualized using an enhanced chemiluminescence kit (Pierce, Thermo Fisher Scientific, Rockford, IL, USA). Anti-AMP-activated protein kinase (AMPK), anti-Thr172 pAMPK, anti-acetyl-CoA-carboxylase (ACC), anti-Ser79 ACC and anti-rabbit secondary were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was purchased from Millipore (Billerica, MA, USA) and anti-mouse secondary was purchased from Santa Cruz Biotechnologies (Dallas, TX, USA).
Targeted tracer fate association study
PC-3 cells (2 × 106) maintained in RPMI medium supplemented with 10% FBS were seeded into 100 mm dishes in triplicate in glucose-free RPMI (Gibco) supplemented with 10% FBS containing 3 mM [1,2-13C2]-D-glucose tracer and 3 mM glucose (Sigma, St. Louis, MO, USA), in the presence or absence of 100 mM methotrexate. Cells were placed into a 37 °C incubator with 5% CO2. Cell pellets were collected after 24 h for analysis. Briefly, culture media was remove, cells were rinsed with 37 °C PBS and then 2 ml 0.25% trypsin-EDTA was added for 3 min or until the cells detached. Cells were spun at 1200 r.p.m. for 5 min. Cell pellets and supernatant (10 ml from each cell line) were frozen at −80 °C in 15 ml tubes.
Flux rates were determined using SiDMAP’s tracer-substrate-based GC–MS targeted tracer fate association EZTopolome platform.58 The analyses include determining associations and their response to MTX treatment via correlations among positional 13C tracer enrichment patterns in multiple intermediary metabolites of glycolysis, glycogen synthesis, tricarboxylic acid cycle, citrate shuttling, fatty acid de novo synthesis, chain elongation and desaturation, gluconeogenesis, carbon exchange in the pentose cycle, glucose-6P-dehydrogenase flux as well as complete glucose oxidation into 13CO2. The retention times and mass-to-charge (m/z) ion clusters of selected ions of cells and culture media metabolites were determined using mass isotopomer distribution analysis (MIDA)59 expressed as net fluxes by subtracting reverse fluxes from forward tracer incorporation patterns via reversible metabolic steps. Results are expressed as peak area. Each experiment was carried out using triplicate cell cultures (a total of 12 samples). Mass spectroscopic analyses were carried out by three independent automatic injections of 1 μl samples by the automatic sampler and accepted only if the standard sample deviation were less than 1% of the normalized peak intensity. For reversible carbon exchange reactions both forward and reverse fluxes were determined and used to calculate net fluxes toward product synthesis.
Abbreviations
- SOG:
-
serine synthesis, one-carbon metabolism and the glycine cleavage system
- GC:
-
glycine cleavage
- OxPhos:
-
oxidative phosphorylation
- TTFAS:
-
targeted tracer fate association study
- PKM2:
-
pyruvate kinase M2 isoform
- GSEA:
-
gene set enrichment analysis
- PCC:
-
Pearson's correlation coefficient
- GLUD:
-
glutamate dehydrogenase
- G6PD:
-
glucose-6-phosphate dehydrogenase
- FAS:
-
fatty acid synthase
- AMPK:
-
AMP kinase
- ACC:
-
acetyl-CoA carboxylase
- pAMPK:
-
phosphorylated AMPK
- pACC:
-
phosphorylated ACC
- MTX:
-
methotrexate
- ALL:
-
acute lymphocytic leukemia
References
Vander Heiden MG, Cantley LC, Thompson CB . Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033.
Munoz-Pinedo C, El Mjiyad N, Ricci JE . Cancer metabolism: current perspectives and future directions. Cell Death Dis 2012; 3: e248.
Zhao Y, Butler EB, Tan M . Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013; 4: e532.
Warburg O . On the origin of cancer cells. Science 1956; 123: 309–314.
Kit S . The biosynthesis of free glycine and serine by tumors. Cancer Res 1955; 15: 715–718.
Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011; 476: 346–350.
Pollari S, Käkönen SM, Edgren H, Wolf M, Kohonen P, Sara H et al. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res Treat 2011; 125: 421–430.
Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 2011; 43: 869–874.
Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012; 148: 259–272.
Dominy JE, Vazquez F, Puigserver P . Glycine decarboxylase cleaves a ‘malignant’ metabolic path to promote tumor initiation. Cancer Cell 2012; 21: 143–145.
Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012; 336: 1040–1044.
Vazquez A, Liu J, Zhou Y, Oltvai ZN . Catabolic efficiency of aerobic glycolysis: The Warburg effect revisited. BMC Syst Biol 2010; 4: 58.
Vazquez A, Oltvai ZN . Molecular crowding defines a common origin for the warburg effect in proliferating cells and the lactate threshold in muscle physiology. Plos One 2011; 6: e19538.
Vazquez A, Markert EK, Oltvai ZN . Serine biosynthesis with one carbon catabolism and the glycine cleavage system represents a novel pathway for ATP generation. Plos One 2011; 6: e25881.
Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010; 329: 1492–1499.
Mazurek S . Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol 2011; 43: 969–980.
Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest 2013; 123: 2921–2934.
Yang Y, Lane AN, Ricketts CJ, Sourbier C, Wei MH, Shuch B et al. Metabolic reprogramming for producing energy and reducing power in Fumarate Hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PloS One 2013; 8: e72179.
Hum DW, Bell AW, Rozen R, MacKenzie RE . Primary structure of a human trifunctional enzyme. Isolation of a cDNA encoding methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase-formyltetrahydrofolate synthetase. J Biol Chem 1988; 263: 15946–15950.
Mejia NR, MacKenzie RE . NAD-dependent methylenetetrahydrofolate dehydrogenase is expressed by immortal cells. J Biol Chem 1985; 260: 14616–14620.
Bolusani S, Young BA, Cole NA, Tibbetts AS, Momb J, Bryant JD et al. Mammalian MTHFD2L encodes a mitochondrial methylenetetrahydrofolate dehydrogenase isozyme expressed in adult tissues. J Biol Chem 2011; 286: 5166–5174.
Prasannan P, Pike S, Peng K, Shane B, Appling DR . Human mitochondrial C1-tetrahydrofolate synthase: gene structure, tissue distribution of the mRNA, and immunolocalization in Chinese hamster ovary calls. J Biol Chem 2003; 278: 43178–43187.
Locasale JW . Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer 2013; 13: 572–583.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550.
Stuart-Harris R, Caldas C, Pinder SE, Pharoah P . Proliferation markers and survival in early breast cancer: a systematic review and meta-analysis of 85 studies in 32 825 patients. Breast 2008; 17: 323–334.
Chandriani S, Frengen E, Cowling VH, Pendergrass SA, Perou CM, Whitfield ML et al. A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. Plos One 2009; 4: e6693.
Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PE, Nelson SJ et al. (13)C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc-induced tumor formation and regression. Cell Metab 2011; 14: 131–142.
Santarius T, Shipley J, Brewer D, Stratton MR, Cooper CS . A census of amplified and overexpressed human cancer genes. Nat Rev Cancer 2010; 10: 59–64.
Cairns RA, Harris IS, Mak TW . Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11: 85–95.
Lu SJ, Hipp JA, Feng Q, Hipp JD, Lanza R, Atala A . GeneChip analysis of human embryonic stem cell differentiation into hemangioblasts: an in silico dissection of mixed phenotypes. Genome Biol 2007; 8: R240.
Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV . An integrated database of genes responsive to the Myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol 2003; 4: R69.
Hopfl G, Gassmann M, Desbaillets I . Differentiating embryonic stem cells into embryoid bodies. Methods Mol Biol 2004; 254: 79–98.
Ciszak EM, Makal A, Hong YS, Vettaikkorumakankauv AK, Korotchkina LG, Patel MS . How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J Biol Chem 2006; 281: 648–655.
McCartney RG, Rice JE, Sanderson SJ, Bunik V, Lindsay H, Lindsay JG . Subunit interactions in the mammalian alpha-ketoglutarate dehydrogenase complex. Evidence for direct association of the alpha-ketoglutarate dehydrogenase and dihydrolipoamide dehydrogenase components. J Biol Chem 1998; 273: 24158–24164.
Conacci-Sorrel M, Ngouenet C, Eisenman RN . Myc-nick: a cytoplasmic cleavage product of Myc that promotes alpha-tubulin acetylation and cell differentiation. Cell 2010; 142: 480–493.
Moghaddas Gholami A, Hahne H, Wu Z, Auer FJ, Meng C, Wilhelm M et al. Global proteome analysis of the NCI-60 cell line panel. Cell Rep 2013; 4: 609–620.
Schellenberger J, Park JO, Conrad TM, Palsson BO . BiGG: a biochemical genetic and genomic knowledge base of large scale metabolic reconstructions. BMC Bioinformatics 2010; 11: 213.
Gerosa L, Sauer U . Regulation and control of metabolic fluxes in microbes. Curr Opin Biotechnol 2011; 22: 566–575.
Snell K, Fell DA . Metabolic control analysis of mammalian serine metabolism. Adv Enzyme Regul 1990; 30: 13–32.
Hardie DG . AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 2011; 25: 1895–1908.
Metallo CM, Walther JL, Stephanopoulos G . Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J Biotechnol 2009; 144: 167–174.
Allegra CJ, Fine RL, Drake JC, Chabner BA . The effect of methotrexate on intracellular folate pools in human MCF-7 breast cancer cells. Evidence for direct inhibition of purine synthesis. J Biol Chem 1986; 261: 6478–6485.
McGuire JJ . Anticancer antifolates: current status and future directions. Curr Pharm Des 2003; 9: 2593–2613.
Vazquez A, Tedeschi PM, Bertino JR . Overexpression of the mitochondrial folate and glycine-serine pathway: a new determinant of methotrexate selectivity in tumors. Cancer Res 2013; 73: 478–482.
Tibbetts AS, Appling DR . Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr 2010; 30: 57–81.
Fu TF, Rife JP, Schirch V . The role of serine hydroxymethyltransferase isozymes in one-carbon metabolism in MCF-7 cells as determined by (13)C NMR. Arch Biochem Biophys 2001; 393: 42–50.
Horne DW, Holloway RS, Said HM . Uptake of 5-formyltetrahydrofolate in isolated rat liver mitochondria is carrier-mediated. J Nutr 1992; 122: 2204–2209.
Lawrence SA, Hackett JC, Moran RG . Tetrahydrofolate recognition by the mitochondrial folate transporter. J Biol Chem 2011; 286: 31480–31489.
van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002; 415: 530–536.
van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002; 347: 1999–2009.
Sboner A, Demichelis F, Calza S, Pawitan Y, Setlur SR, Hoshida Y et al. Molecular sampling of prostate cancer: a dilemma for predicting disease progression. BMC Med Genomics 2010; 3: 8.
Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008; 40: 499–507.
Markert EK, Mizuno H, Vazquez A, Levine AJ . Molecular classification of prostate cancer using curated expression signatures. Proc Natl Acad Sci USA 2011; 108: 21276–21281.
Lo Surdo J, Bauer SR . Quantitative approaches to detect donor and passage differences in adipogenic potential and clonogenicity in human bone marrow-derived mesenchymal stem cells. Tissue Eng Part C Methods 2012; 18: 877–889.
Chan LL, Wilkinson AR, Paradis BD, Lai N . Rapid image-based cytometry for comparison of fluorescent viability staining methods. J Fluoresc 2012; 22: 1301–1311.
Chan LL, Zhong X, Qiu J, Li PY, Lin B . Cellometer vision as an alternative to flow cytometry for cell cycle analysis, mitochondrial potential, and immunophenotyping. Cytometry A 2011; 79: 507–517.
Bhatt DP, Chen X, Geiger JD, Rosenberger TA . A sensitive HPLC-based method to quantify adenine nucleotides in primary astrocyte cell cultures. J Chromatogr B Analyt Technol Biomed Life Sci 2012; 889-890: 110–115.
Cantoria MJ, Boros LG, Meuillet MJ . Contextual inhibition of fatty acid synthesis by metformin involves glucose-derived acetyl-CoA and cholesterol in pancreatic tumor cells. Metabolomics 2013 doi:10.1007/s11306-013-0555-4.
Lee WN . Stable isotopes and mass isotopomer study of fatty acid and cholesterol synthesis. A review of the MIDA approach. Adv Exp Med Biol 1996; 399: 95–114.
Acknowledgements
This work was funded by the RWJ Foundation and NCI P30 CA072720.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
LL-YC and JQ declare a financial interest in the image cytometry system used in the manuscript. The authors LL-YC and JQ are employees of Nexcelom Bioscience, LL-YC, who is the manufacturer of the Cellometer Auto T4 image cytometer utilized in the manuscript.
Additional information
Edited by G Melino
Author Contributions
ZNO and AV conceived the work. EKM and AV performed the gene expression analysis and the metabolic model simulations. PMT and DD conducted the SOG pathway inhibition experiments under the supervision of JRB and RSD. MG and HL performed the adenine nucleotide measurements. SCD performed the protein content measurements of the NCI60 cell lines under the supervision of KMH. LL-YC and JQ performed the cell size measurements of the NCI60 cell lines. LGB contributed with the flux measurements performed at SiDMAP. All authors contributed to writing the manuscript.
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Tedeschi, P., Markert, E., Gounder, M. et al. Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis 4, e877 (2013). https://doi.org/10.1038/cddis.2013.393
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.393
Keywords
This article is cited by
-
Unraveling the role of the mitochondrial one-carbon pathway in undifferentiated thyroid cancer by multi-omics analyses
Nature Communications (2024)
-
Targeting serine-glycine-one-carbon metabolism as a vulnerability in cancers
Biomarker Research (2023)
-
Bayesian kinetic modeling for tracer-based metabolomic data
BMC Bioinformatics (2023)
-
AMPK-HIF-1α signaling enhances glucose-derived de novo serine biosynthesis to promote glioblastoma growth
Journal of Experimental & Clinical Cancer Research (2023)
-
Optimal energy and redox metabolism in the cyanobacterium Synechocystis sp. PCC 6803
npj Systems Biology and Applications (2023)
